
Influence of Polymorphism on the Electrochemical Behavior of Dilithium (2,3-Dilithium-oxy)-terephthalate vs. Li

 , and
, and 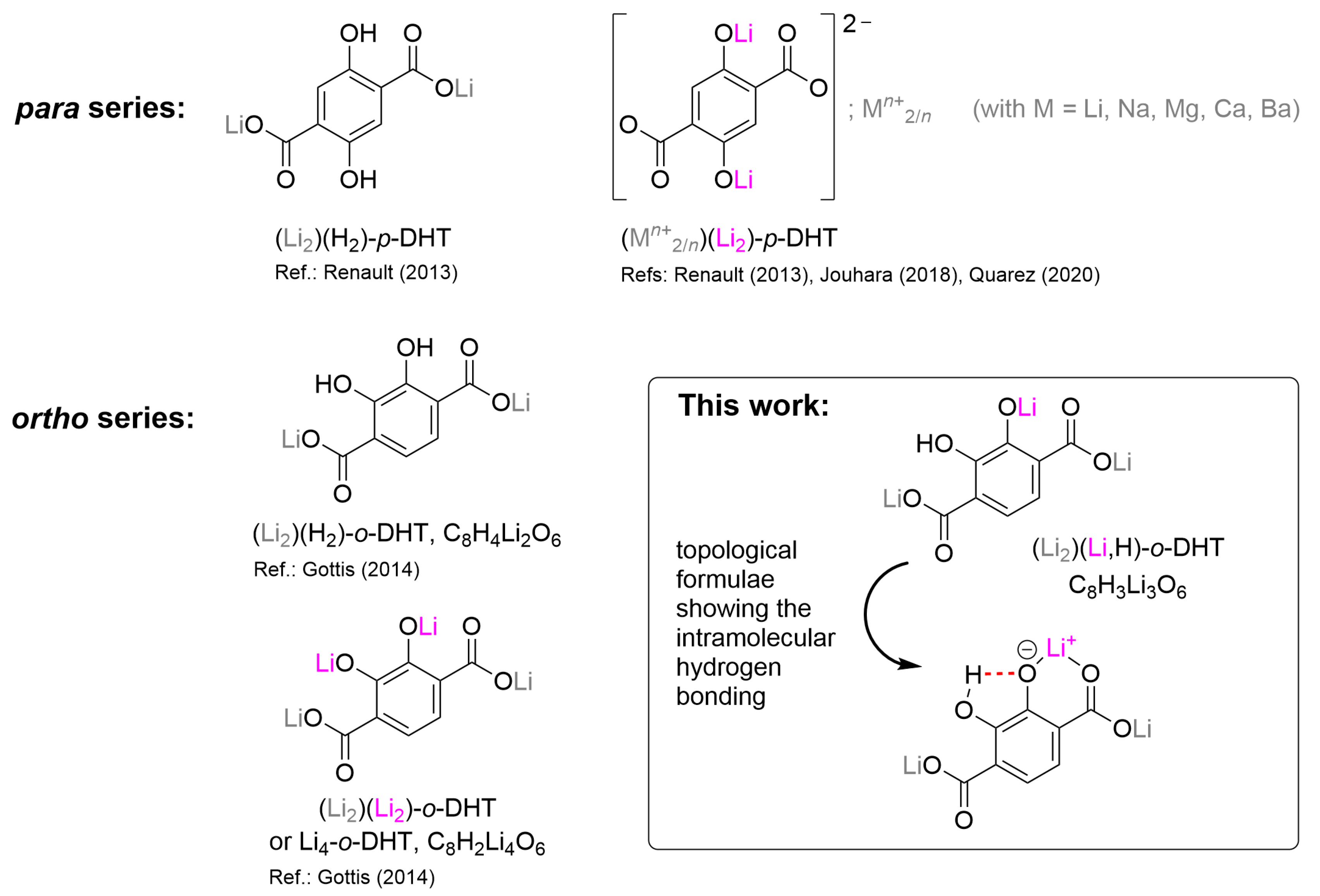{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Preliminary Observations
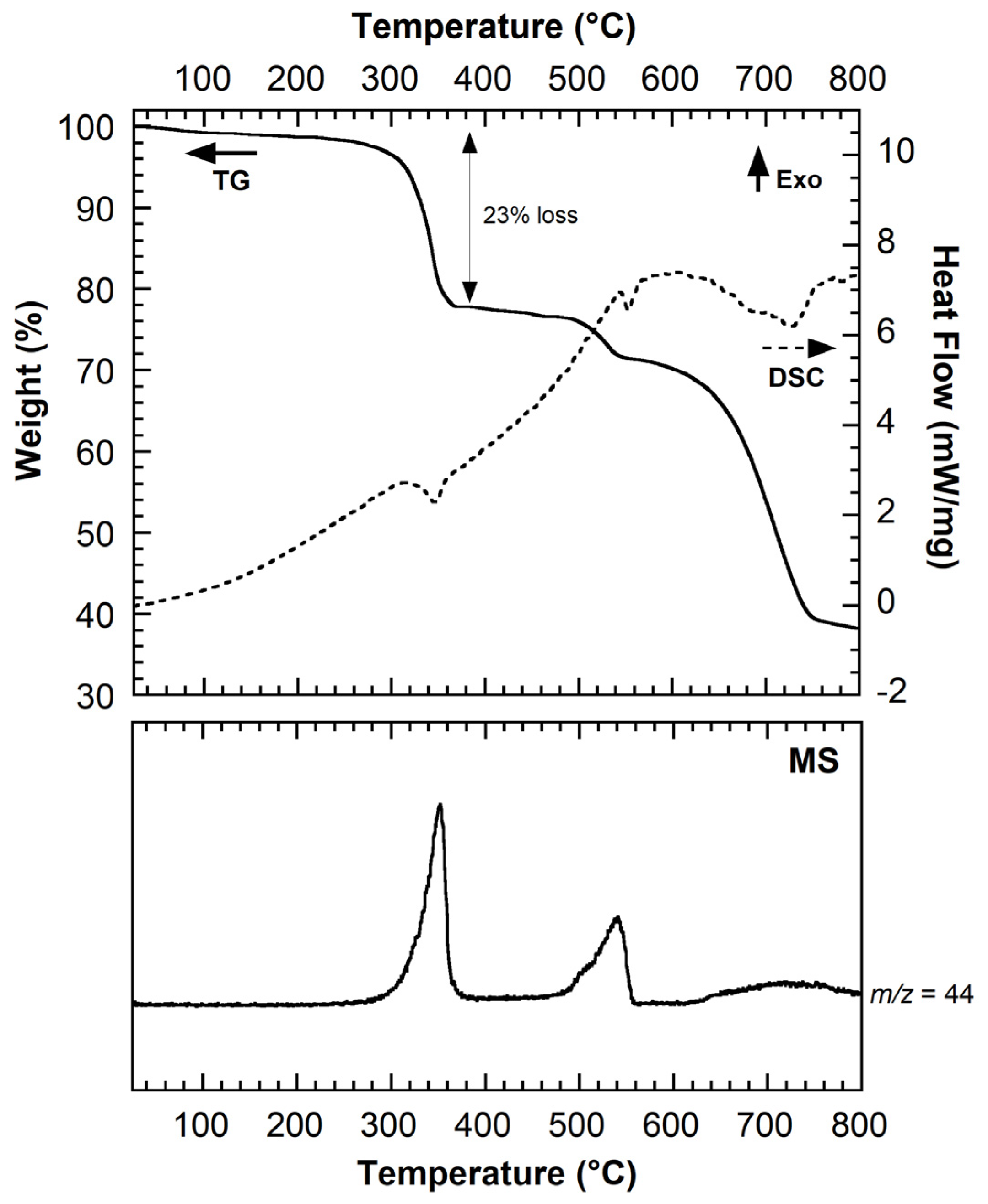
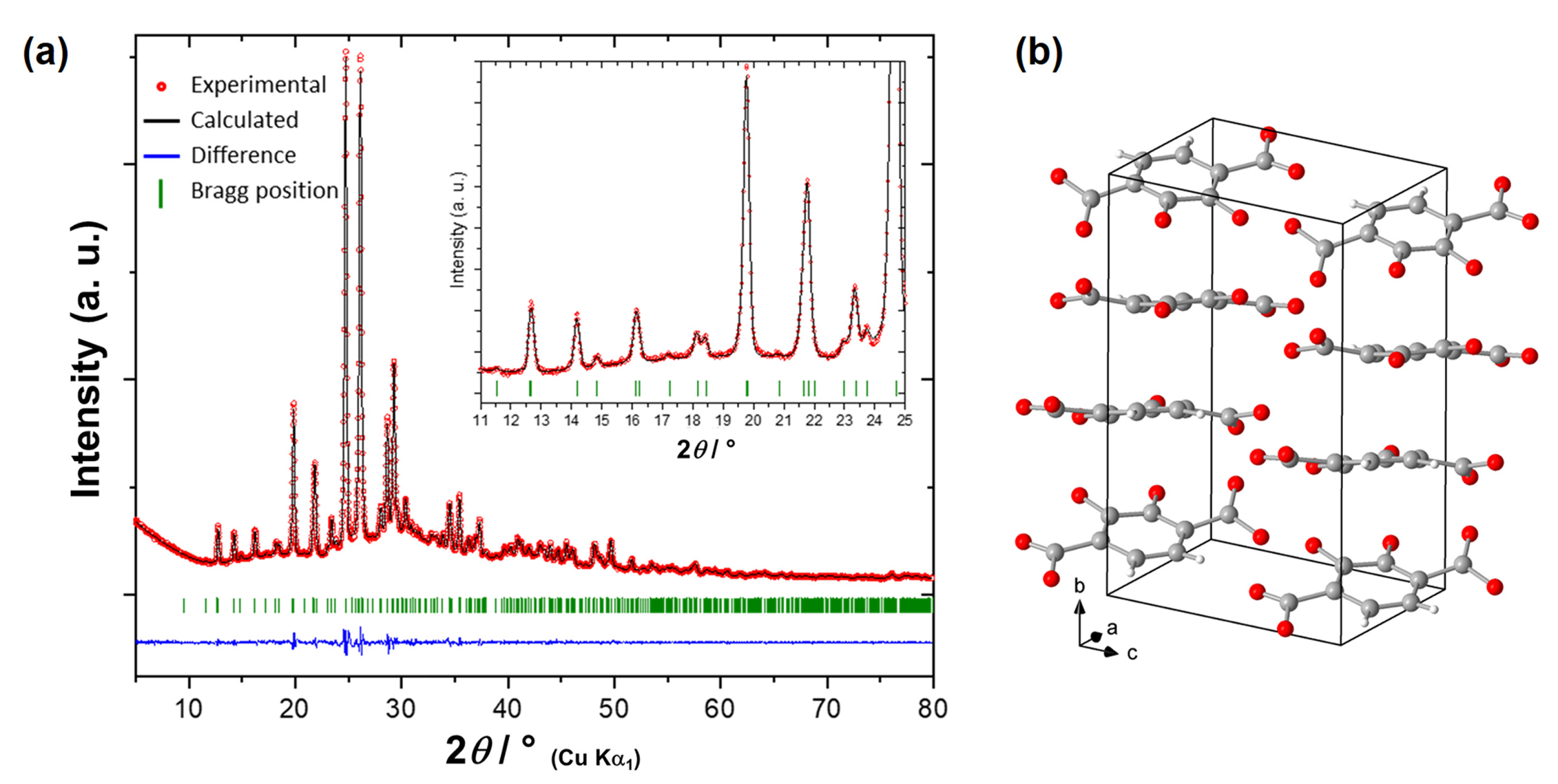
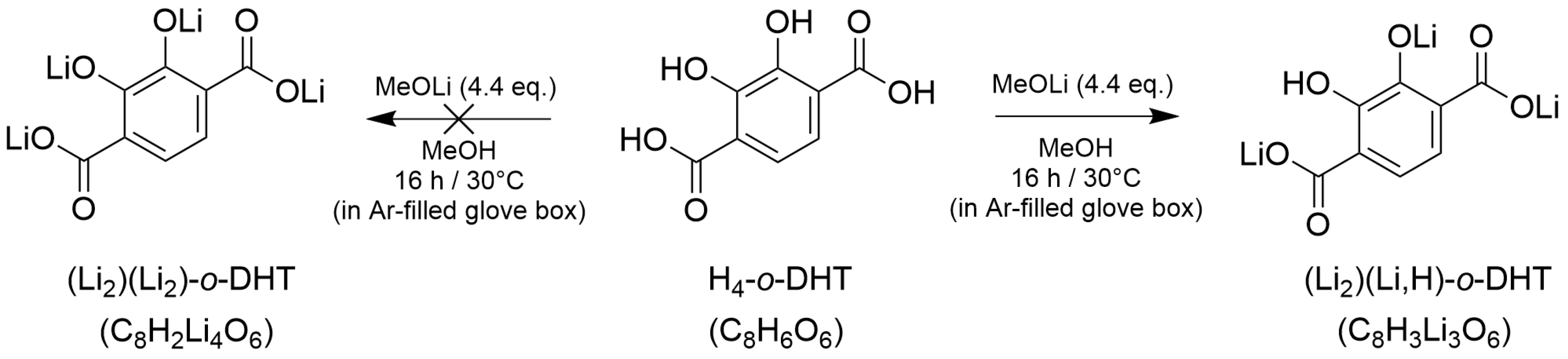
2.2. Synthesis, Characterizations and Thermal Behavior of (Li2)(Li,H)-o-DHT
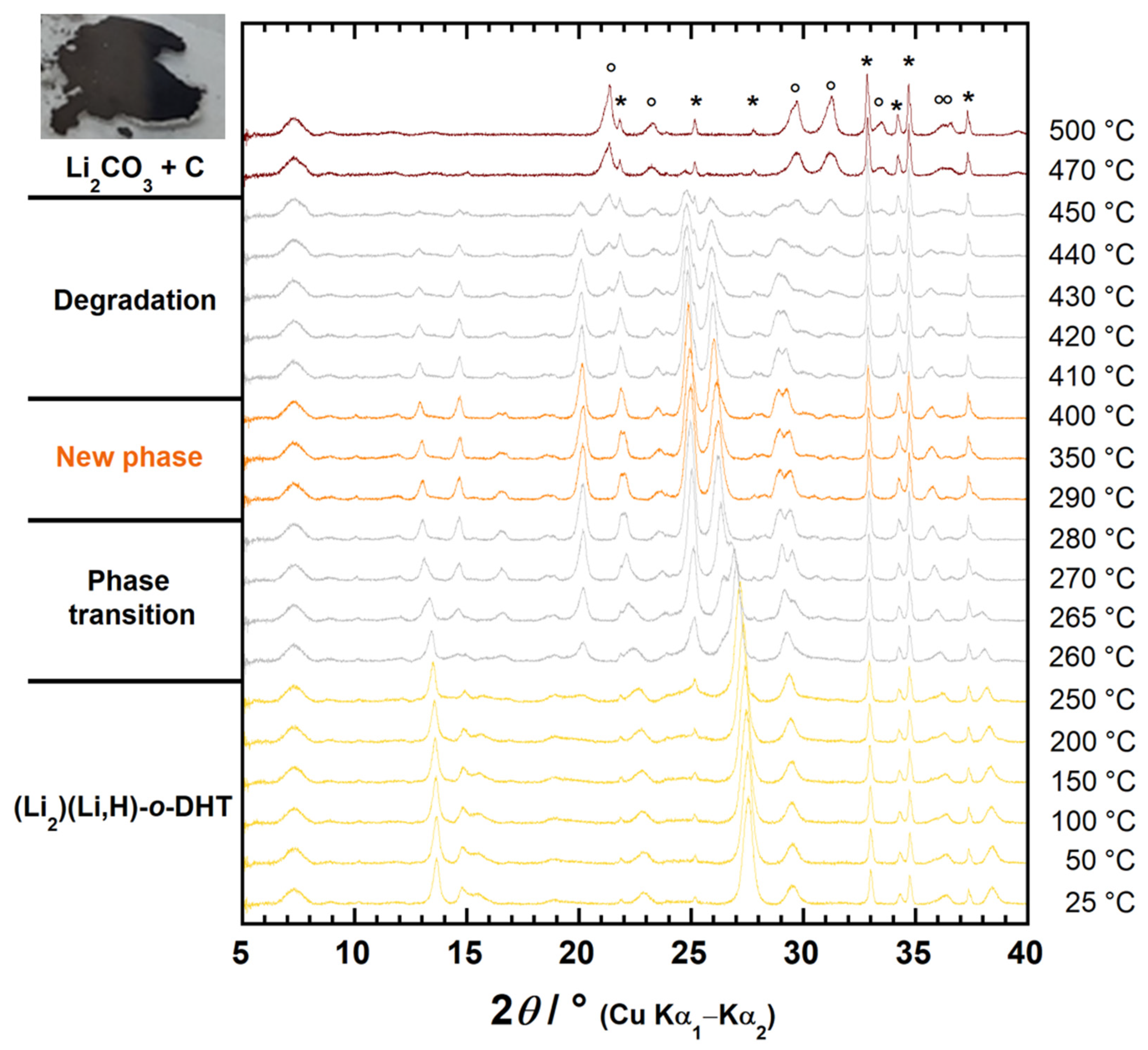
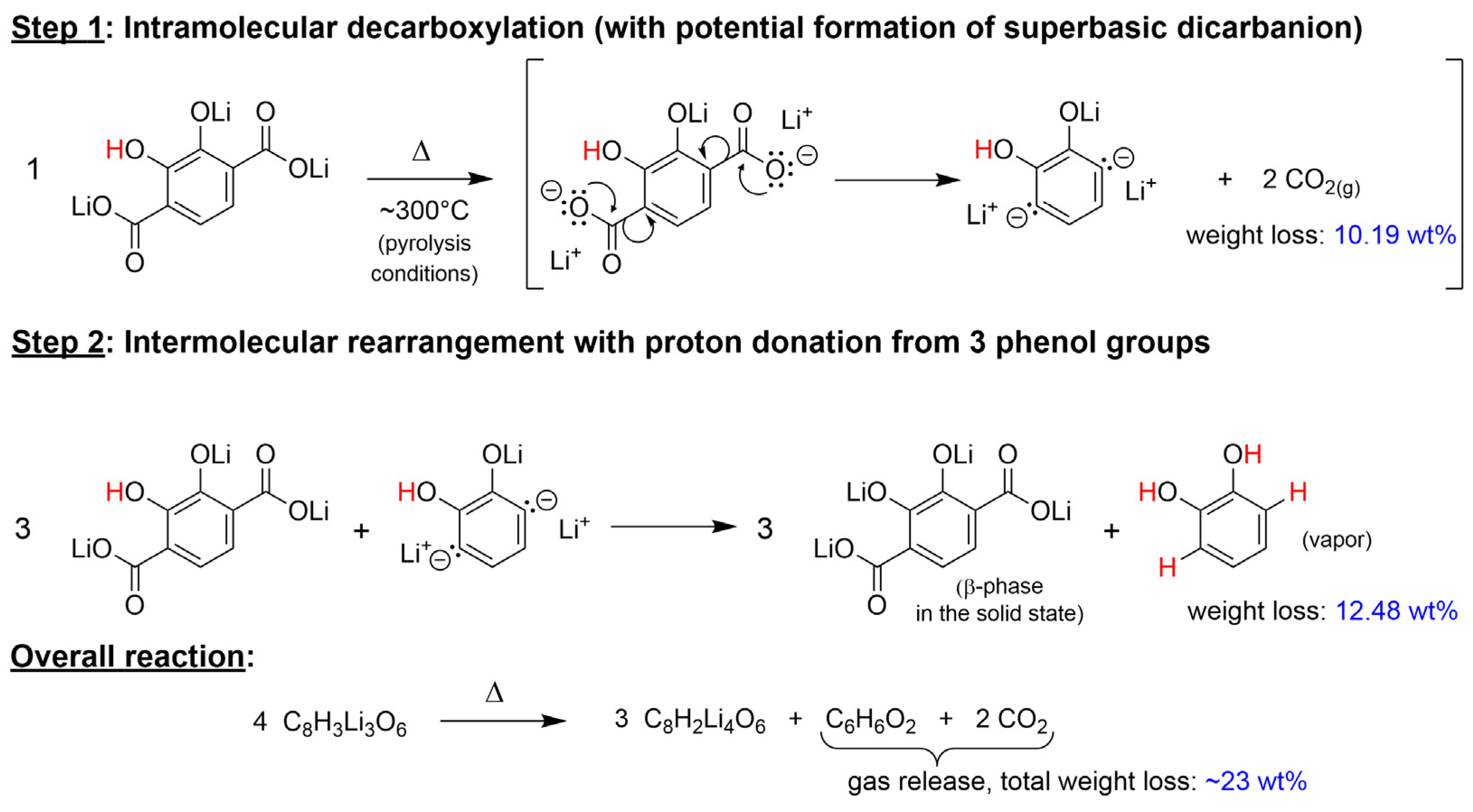
2.3. Identification of the New Phase and Proposed Thermal Rearrangement Mechanism
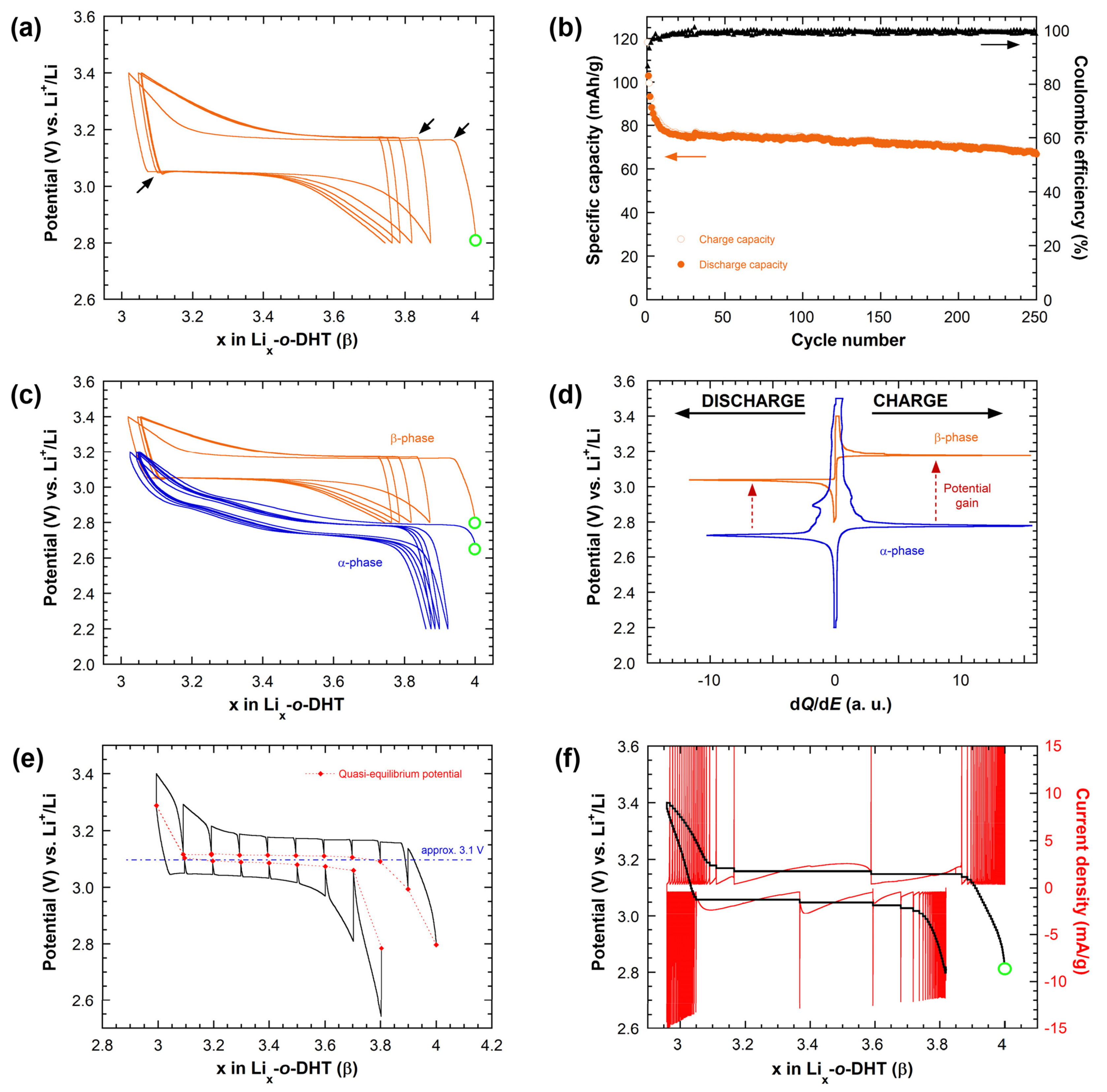
2.4. Electrochemical Behavior of Li4-o-DHT (β-Phase) as Active Electrode Material vs. Li
2.5. Redox Potential Tuning in the Solid State: From Polymorphism to Through-Space Charge Modulation
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gulley, A.L.; McCullough, E.A.; Shedd, K.B. China’s Domestic and Foreign Influence in the Global Cobalt Supply Chain. Resour. Policy 2019, 62, 317–323. [Google Scholar] [CrossRef]
- Petavratzi, E.; Gunn, G.; Kresse, C. BGS Commodity Review: Cobalt; British Geological Survey: Nottingham, UK, 2019. [Google Scholar]
- Baars, J.; Domenech, T.; Bleischwitz, R.; Melin, H.E.; Heidrich, O. Circular Economy Strategies for Electric Vehicle Batteries Reduce Reliance on Raw Materials. Nat. Sustain. 2021, 4, 71–79. [Google Scholar] [CrossRef]
- Curtis, T.; Smith, L.; Buchanan, H.; Heath, G. A Circular Economy for Lithium-Ion Batteries Used in Mobile and Stationary Energy Storage: Drivers, Barriers, Enablers, and U.S. Policy Considerations; National Renewable Energy Lab. (NREL): Golden, CO, USA, 2021; p. NREL/TP--6A20-77035. [Google Scholar]
- Sommerville, R.; Zhu, P.; Rajaeifar, M.A.; Heidrich, O.; Goodship, V.; Kendrick, E. A Qualitative Assessment of Lithium Ion Battery Recycling Processes. Resour. Conserv. Recycl. 2021, 165, 105219. [Google Scholar] [CrossRef]
- Poizot, P.; Dolhem, F. Clean Energy New Deal for a Sustainable World: From Non-CO2 Generating Energy Sources to Greener Electrochemical Storage Devices. Energy Environ. Sci. 2011, 4, 2003–2019. [Google Scholar] [CrossRef]
- Poizot, P.; Gaubicher, J.; Renault, S.; Dubois, L.; Liang, Y.; Yao, Y. Opportunities and Challenges for Organic Electrodes in Electrochemical Energy Storage. Chem. Rev. 2020, 120, 6490–6557. [Google Scholar] [CrossRef]
- Esser, B.; Dolhem, F.; Becuwe, M.; Poizot, P.; Vlad, A.; Brandell, D. A Perspective on Organic Electrode Materials and Technologies for next Generation Batteries. J. Power Sources 2021, 482, 228814. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, Q. Recent Progress in Aqueous Monovalent-Ion Batteries with Organic Materials as Promising Electrodes. Mater. Today Energy 2020, 18, 100547. [Google Scholar] [CrossRef]
- Esser, B. Redox Polymers as Electrode-Active Materials for Batteries. Org. Mater. 2019, 1, 063–070. [Google Scholar] [CrossRef] [Green Version]
- Saal, A.; Hagemann, T.; Schubert, U.S. Polymers for Battery Applications—Active Materials, Membranes, and Binders. Adv. Energy Mater. 2021, 11, 2001984. [Google Scholar] [CrossRef]
- Dühnen, S.; Betz, J.; Kolek, M.; Schmuch, R.; Winter, M.; Placke, T. Toward Green Battery Cells: Perspective on Materials and Technologies. Small Methods 2020, 4, 2000039. [Google Scholar] [CrossRef]
- Lakraychi, A.E.; Dolhem, F.; Vlad, A.; Becuwe, M. Organic Negative Electrode Materials for Metal-Ion and Molecular-Ion Batteries: Progress and Challenges from a Molecular Engineering Perspective. Adv. Energy Mater. 2021, 11, 2101562. [Google Scholar] [CrossRef]
- Lu, Y.; Cai, Y.; Zhang, Q.; Chen, J. Structure–Performance Relationships of Covalent Organic Framework Electrode Materials in Metal-Ion Batteries. J. Phys. Chem. Lett. 2021, 12, 8061–8071. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, K.; Yamauchi, Y.; Oyaizu, K.; Jia, Z. Nitroxide Radical Polymers for Emerging Plastic Energy Storage and Organic Electronics: Fundamentals, Materials, and Applications. Mater. Horiz. 2021, 8, 803–829. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lakraychi, A.E.; Chen, Z.; Liang, Y.; Yao, Y. Roadmap of Solid-State Lithium-Organic Batteries toward 500 Wh Kg−1. ACS Energy Lett. 2021, 6, 3287–3306. [Google Scholar] [CrossRef]
- Ravet, N.; Michot, C.; Armand, M. Novel Cathode Materials Based on Organic Couples for Lithium Batteries. MRS Proc. 1997, 496, 263. [Google Scholar] [CrossRef]
- Renault, S.; Gottis, S.; Barrès, A.-L.; Courty, M.; Chauvet, O.; Dolhem, F.; Poizot, P. A Green Li–Organic Battery Working as a Fuel Cell in Case of Emergency. Energy Environ. Sci. 2013, 6, 2124–2133. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Chen, C.; Ma, T.; Chen, J. Nanostructured Organic Electrode Materials Grown on Graphene with Covalent-Bond Interaction for High-Rate and Ultra-Long-Life Lithium-Ion Batteries. Nano Res. 2017, 10, 4245–4255. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Zhang, K.; Zhu, Z.; Tao, Z.; Chen, J. Organic Li4C8H2O6 Nanosheets for Lithium-Ion Batteries. Nano Lett. 2013, 13, 4404–4409. [Google Scholar] [CrossRef]
- Gottis, S.; Barrès, A.-L.; Dolhem, F.; Poizot, P. Voltage Gain in Lithiated Enolate-Based Organic Cathode Materials by Isomeric Effect. ACS Appl. Mater. Interfaces 2014, 6, 10870–10876. [Google Scholar] [CrossRef]
- Miao, L.; Liu, L.; Shang, Z.; Li, Y.; Lu, Y.; Cheng, F.; Chen, J. The Structure–Electrochemical Property Relationship of Quinone Electrodes for Lithium-Ion Batteries. Phys. Chem. Chem. Phys. 2018, 20, 13478–13484. [Google Scholar] [CrossRef]
- Jouhara, A.; Dupré, N.; Gaillot, A.-C.; Guyomard, D.; Dolhem, F.; Poizot, P. Raising the Redox Potential in Carboxyphenolate-Based Positive Organic Materials via Cation Substitution. Nat. Commun. 2018, 9, 4401. [Google Scholar] [CrossRef] [Green Version]
- Quarez, É.; Jouhara, A.; Grolleau, S.; Dolhem, F.; Dupré, N.; Poizot, P. From Partial to Complete Neutralization of 2,5-Dihydroxyterephthalic Acid in the Li–Na System: Crystal Chemistry and Electrochemical Behavior of Na2Li2C8H2O6 vs. Li. CrystEngComm 2020, 22, 1653–1663. [Google Scholar] [CrossRef]
- Chen, H.; Armand, M.; Courty, M.; Jiang, M.; Grey, C.P.; Dolhem, F.; Tarascon, J.-M.; Poizot, P. Lithium Salt of Tetrahydroxybenzoquinone: Toward the Development of a Sustainable Li-Ion Battery. J. Am. Chem. Soc. 2009, 131, 8984–8988. [Google Scholar] [CrossRef]
- Neng, N.R.; Nogueira, J.M.F. Monitoring Trace Levels of Hydroxy Aromatic Compounds in Urine Matrices by Bar Adsorptive Microextraction (BAμE). Anal. Methods 2017, 9, 5260–5265. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Wang, C.-H.; Liang, H. Scales of Oxidation Potentials, pKa, and BDE of Various Hydroquinones and Catechols in DMSO. J. Org. Chem. 2010, 75, 7240–7257. [Google Scholar] [CrossRef]
- Wesołowski, M. Thermal Decomposition of Salicylic Acid and Its Salts. Thermochim. Acta 1979, 31, 133–146. [Google Scholar] [CrossRef]
- Wenger, M.; Armbruster, T. Crystal Chemistry of Lithium; Oxygen Coordination and Bonding. Eur. J. Mineral. 1991, 3, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Gaubicher, J.; Chabre, Y.; Angenault, J.; Lautié, A.; Quarton, M. Lithium Electrochemical Intercalation in β-VOSO4. J. Alloys Compd. 1997, 262–263, 34–38. [Google Scholar] [CrossRef]
- Ellis, L.D.; Hatchard, T.D.; Obrovac, M.N. Reversible Insertion of Sodium in Tin. J. Electrochem. Soc. 2012, 159, A1801–A1805. [Google Scholar] [CrossRef]
- Meethong, N.; Kao, Y.-H.; Carter, W.C.; Chiang, Y.-M. Comparative Study of Lithium Transport Kinetics in Olivine Cathodes for Li-Ion Batteries. Chem. Mater. 2010, 22, 1088–1097. [Google Scholar] [CrossRef]
- Thackeray, M.M. Manganese Oxides for Lithium Batteries. Prog. Solid State Chem. 1997, 25, 1–71. [Google Scholar] [CrossRef]
- Dambournet, D.; Belharouak, I.; Amine, K. Tailored Preparation Methods of TiO2 Anatase, Rutile, Brookite: Mechanism of Formation and Electrochemical Properties. Chem. Mater. 2010, 22, 1173–1179. [Google Scholar] [CrossRef]
- Griffith, K.J.; Forse, A.C.; Griffin, J.M.; Grey, C.P. High-Rate Intercalation without Nanostructuring in Metastable Nb2O5 Bronze Phases. J. Am. Chem. Soc. 2016, 138, 8888–8899. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Liu, T.; Yu, H.; Ran, F.; Ye, W.; Zhu, H.; Shui, M.; Xie, Y.; Shu, J. Polymorphism-Controlled Electrochemical Energy Storage Performance of LiNbWO6. Chem. Mater. 2020, 32, 3376–3384. [Google Scholar] [CrossRef]
- Rousse, G.; Tarascon, J.M. Sulfate-Based Polyanionic Compounds for Li-Ion Batteries: Synthesis, Crystal Chemistry, and Electrochemistry Aspects. Chem. Mater. 2014, 26, 394–406. [Google Scholar] [CrossRef]
- Rambabu, D.; Lakraychi, A.E.; Wang, J.; Sieuw, L.; Gupta, D.; Apostol, P.; Chanteux, G.; Goossens, T.; Robeyns, K.; Vlad, A. An Electrically Conducting Li-Ion Metal–Organic Framework. J. Am. Chem. Soc. 2021, 143, 11641–11650. [Google Scholar] [CrossRef]
- Shimizu, A.; Tsujii, Y.; Kuramoto, H.; Nokami, T.; Inatomi, Y.; Hojo, N.; Yoshida, J. Nitrogen-Containing Polycyclic Quinones as Cathode Materials for Lithium-Ion Batteries with Increased Voltage. Energy Technol. 2014, 2, 155–158. [Google Scholar] [CrossRef]
- Yokoji, T.; Matsubara, H.; Satoh, M. Rechargeable Organic Lithium-Ion Batteries Using Electron-Deficient Benzoquinones as Positive-Electrode Materials with High Discharge Voltages. J. Mater. Chem. A 2014, 2, 19347–19354. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, J.; Hagler, A.T. Conformational Polymorphism. The Influence of Crystal Structure on Molecular Conformation. J. Am. Chem. Soc. 1978, 100, 673–681. [Google Scholar] [CrossRef]
- Sieuw, L.; Lakraychi, A.E.; Rambabu, D.; Robeyns, K.; Jouhara, A.; Borodi, G.; Morari, C.; Poizot, P.; Vlad, A. Through-Space Charge Modulation Overriding Substituent Effect: Rise of the Redox Potential at 3.35 V in a Lithium-Phenolate Stereoelectronic Isomer. Chem. Mater. 2020, 32, 9996–10006. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Černý, R. FOX, ‘free Objects for Crystallography’: A Modular Approach to Ab Initio Structure Determination from Powder Diffraction. J. Appl. Crystallogr. 2002, 35, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General Features. Z. Krist.-Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Bérar, J.-F.; Baldinozzi, G. Modeling of Line-Shape Asymmetry in Powder Diffraction. J. Appl. Crystallogr. 1993, 26, 128–129. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernard, L.; Jouhara, A.; Quarez, E.; Levieux-Souid, Y.; Le Caër, S.; Tran-Van, P.; Renault, S.; Poizot, P. Influence of Polymorphism on the Electrochemical Behavior of Dilithium (2,3-Dilithium-oxy)-terephthalate vs. Li. Inorganics 2022, 10, 62. https://doi.org/10.3390/inorganics10050062
Bernard L, Jouhara A, Quarez E, Levieux-Souid Y, Le Caër S, Tran-Van P, Renault S, Poizot P. Influence of Polymorphism on the Electrochemical Behavior of Dilithium (2,3-Dilithium-oxy)-terephthalate vs. Li. Inorganics. 2022; 10(5):62. https://doi.org/10.3390/inorganics10050062
Chicago/Turabian StyleBernard, Lou, Alia Jouhara, Eric Quarez, Yanis Levieux-Souid, Sophie Le Caër, Pierre Tran-Van, Stéven Renault, and Philippe Poizot. 2022. "Influence of Polymorphism on the Electrochemical Behavior of Dilithium (2,3-Dilithium-oxy)-terephthalate vs. Li" Inorganics 10, no. 5: 62. https://doi.org/10.3390/inorganics10050062
APA StyleBernard, L., Jouhara, A., Quarez, E., Levieux-Souid, Y., Le Caër, S., Tran-Van, P., Renault, S., & Poizot, P. (2022). Influence of Polymorphism on the Electrochemical Behavior of Dilithium (2,3-Dilithium-oxy)-terephthalate vs. Li. Inorganics, 10(5), 62. https://doi.org/10.3390/inorganics10050062