1. Introduction
In nuclear fuel recycling, spent nuclear fuels are dissolved in HNO
3(aq), and then subjected to separation and purification of fissile materials such as U and Pu. This process is called
reprocessing, and constitutes one of the most important steps in the nuclear fuel cycle [
1]. While the
plutonium uranium redox extraction (PUREX) process, based on solvent extraction, is well-known as a standard approach for this purpose, a variety of other reprocessing methods based on wet chemical and pyrochemical bases have also been proposed. Our group is also developing a new precipitation-based reprocessing called
nuclear fuel materials selective precipitation, NUMAP [
2], where
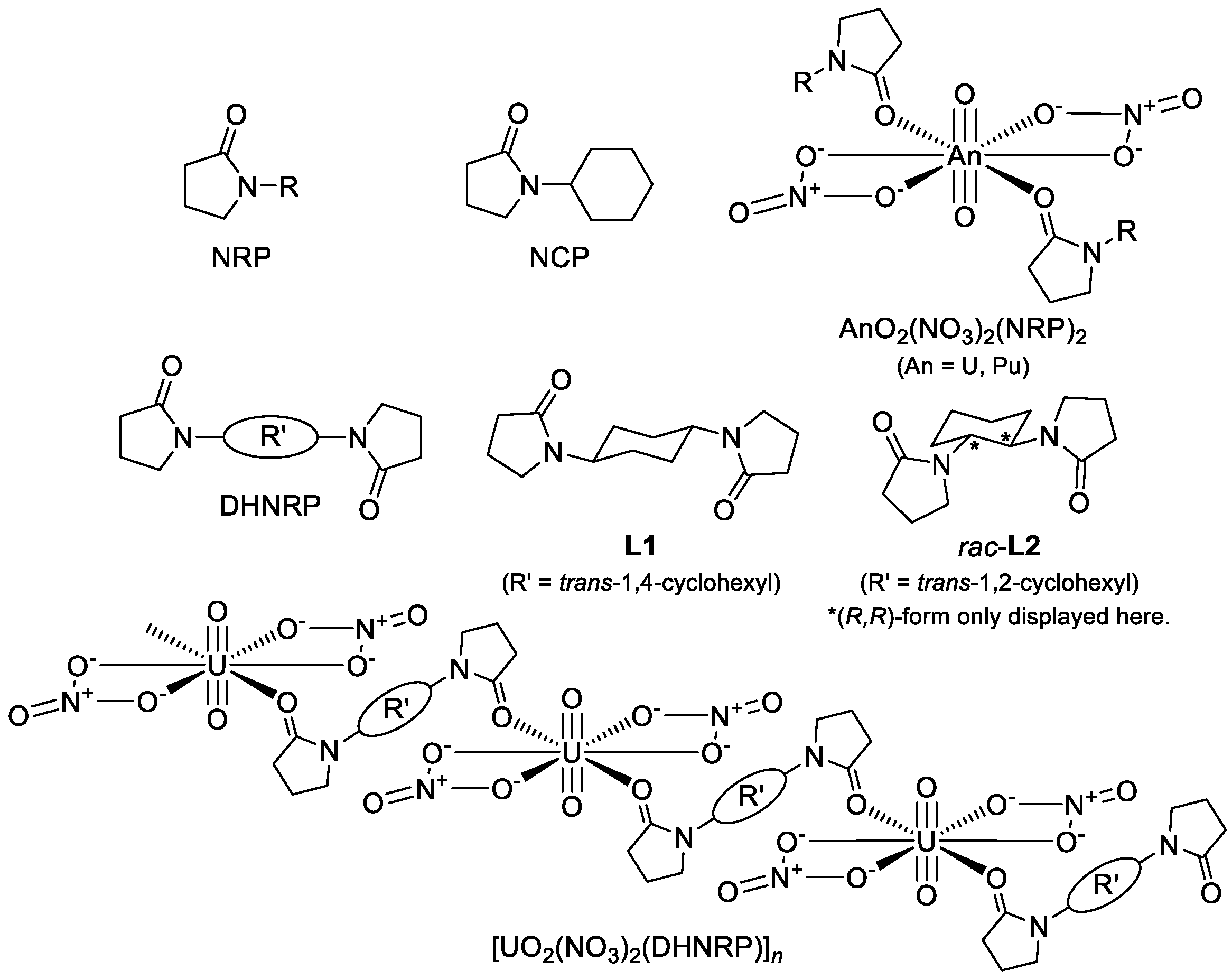
N-alkylated 2-pyrrolidone derivatives (NRPs,
Figure 1) are employed for efficient and simultaneous precipitation of U and Pu from HNO
3(aq) [
3,
4]. In the NUMAP approach, Pu will be deliberately recovered together with U for the sake of securing nuclear proliferation resistivity. This is in strong contrast to PUREX, which has been developed to isolate Pu for nuclear weapons. Similar precipitation-based reprocessing concepts are also proposed by other groups [
5,
6].
Understanding the coordination chemistry of early actinides with NO
3− as well as with selected ligands like NRPs is highly essential to establish foundations of chemical separation in any wet chemical reprocessing processes. The early actinides such as U, Np, and Pu exhibit rich redox chemistry, with common oxidation states varying between III and VI [
7]. In penta- and hexavalent states, actinyl ions (AnO
2m+; An = U, Np, Pu;
m = 1, 2) are formed, where the equatorial plane is offered for additional coordination of extra ligands, as exemplified by AnO
2(NO
3)
2(NRP)
2 in
Figure 1. By exploiting such uniqueness in coordination chemistry, we extended the molecular design of NRPs to double-headed ones, DHNRPs (
Figure 1), to form a much less-soluble [UO
2(NO
3)
2(DHNRP)]
n coordination polymer to further improve the recovery efficiency of UO
22+ precipitate [
8,
9,
10].
In the reprocessing process, Pu is intended to be recovered together with UO
22+, whereas Np should be removed from them. In the first phase of NUMAP development, hydrophobic NRP-like NCP (
Figure 1) has been preferably employed to achieve high recovery yields of UO
22+ and PuO
22+ [
4,
11,
12,
13,
14]. Therefore, we are now strongly motivated to explore how NpO
22+ and PuO
22+, the heavier analogues of UO
22+, react with DHNRPs in HNO
3(aq). While the stability of hexavalent states of Np and Pu are generally decreasing, more common oxidation numbers for them are V and IV, respectively. Nevertheless, we are still interested in determining the coordination behavior of the AnO
22+ family with DHNRPs, through which chemical analogy may be observed. Accordingly, it is valuable to first gain knowledge about what happens on NpO
22+, which is only allowed to be handled in a dedicated facility, in order to judge whether or not it is worth studying PuO
22+ in this direction. Herein, we have assessed the structural chemistry of an NpO
22+ coordination polymer with NO
3− and DHNRPs (
L1,
rac-
L2), as well as its solubility, in 3 M HNO
3(aq), typically used in the reprocessing process.
2. Results and Discussion
A 3 M HNO
3 solution dissolving 100 mM NpO
22+ (25 μL) was layered with a blank 3 M HNO
3(aq) (50 μL) and that containing 200 mM
L1 (12.5 μL) from bottom to top in a glass tube (
ϕ5 mm) inserted into a 2 mL glass vial, where NpO
22+:
L1 = 1:1. This layered mixture was stored in a silent place overnight in a glovebox dedicated for Np experiments. Through natural diffusion of the components in the solution phase, yellowish-brown crystals suitable for X-ray crystallography deposited, as shown in
Figure 2. A γ-ray spectrometry of the supernatant allowed to know that this crystalline deposit contained Np and that its yield was 44% based on the initial loading of NpO
22+.
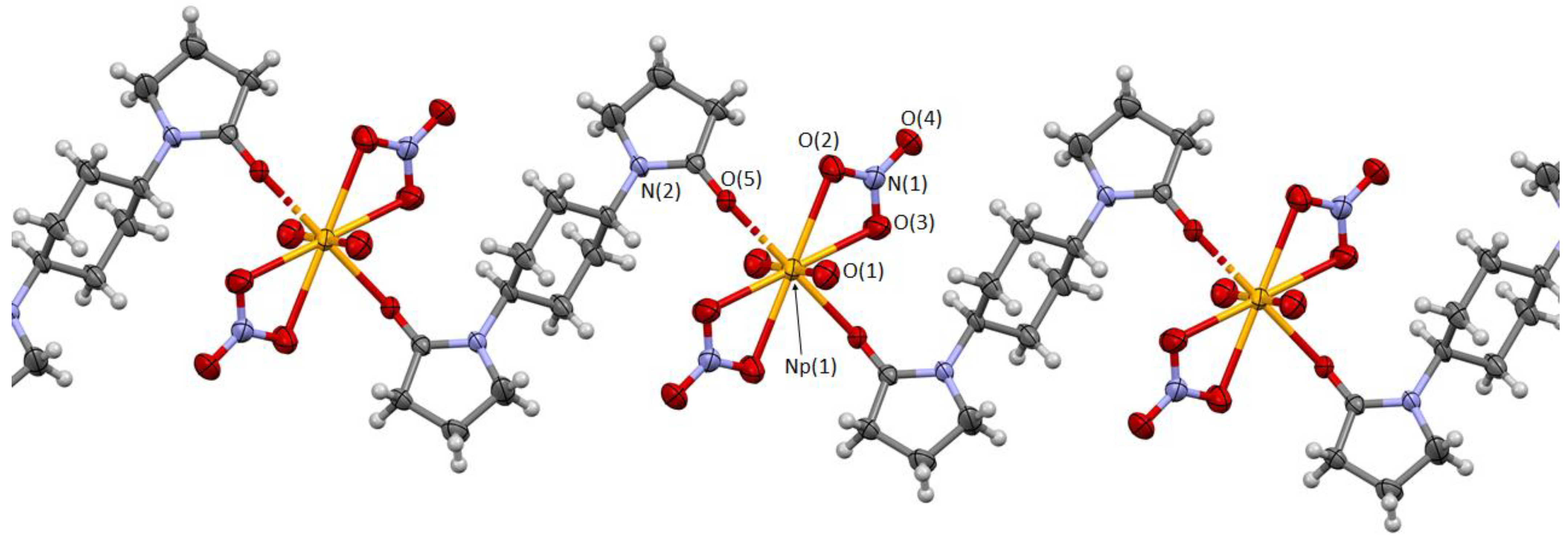
Molecular and crystal structures of this compound were determined by single crystal X-ray analysis at 293 K. As a result, this compound shown in
Figure 3 was found to consist of NpO
2(NO
3)
2 units connected by
L1 linker molecules to form an infinite one-dimensional (1D) coordination polymer, [NpO
2(NO
3)
2(
L1)]
n, which should be formed through the following reaction in the HNO
3 solution.
While we have mentioned above that Np exhibits a wide variety of oxidation numbers from III to VI, or even to VII, as one of unique trends of early actinide elements [
7,
15,
16], its hexavalent oxidation state remained unchanged in the current system.
Crystallographic data of [NpO
2(NO
3)
2(
L1)]
n are summarized in
Table 1. This coordination polymer crystallized in triclinic
P−1 with
a = 6.0172(2) Å,
b = 7.6661(2) Å,
c = 11.0985(3) Å,
α = 75.9330(10)°,
β = 84.488(2)°, and
γ = 76.353(2)°. A volume of its unit cell (
V) was 482.18(2) Å
3. As shown in this table, all of the crystallographic data of the current compound are almost identical to those of its UO
22+ analogue, [UO
2(NO
3)
2(
L1)]
n (triclinic
P–1,
a = 5.8962(4) Å,
b = 7.6330(6) Å,
c = 11.0279(9) Å,
α = 76.841(5)°,
β = 84.480(6)°,
γ = 76.229(5)°, and
V = 468.93(6) Å
3), we reported previously [
9]. Both UO
22+ and NpO
22+ form isomorphous crystalline phases of [AnO
2(NO
3)
2(
L1)]
n because of the chemical similarity of AnO
22+ ions.
As shown in
Figure 3, the central Np atom is surrounded by two axial O atoms of NpO
22+ (O
ax) and by six equatorial O atoms of two bidentate NO
3− ions (O
NO3) and two monodentate 2-pyrrolidone moieties of the different
L1 molecules (O
L) to afford a hexagonal bipyramidal coordination polyhedron. The asymmetric unit comprises only one Np, one O
ax, one NO
3−, and half of
L1, which is expanded by an inversion center (symmetry operation: (i) 1 − x, 1 − y, −z) to complete a whole structure of the [NpO
2(NO
3)
2(
L1)] monomer. As a result, these sets of ligands are located at
trans-positions, which is rather ordinarily found in actinyl(VI) nitrates, as reviewed elsewhere [
17,
18].
Selected structural parameters of [NpO
2(NO
3)
2(
L1)]
n were summarized in
Table 2 together with those of [UO
2(NO
3)
2(
L1)]
n [
9]. The Np≡O
ax bond length (Np(1)−O(1)) is 1.744(3) Å, while longer distances can be found in Np−O
NO3 (Np−O(2), Np−O(3): mean 2.56 Å) and Np−O
L (Np−O(5): 2.341(2) Å) interactions formed in the equatorial plane. The O
ax≡Np≡O
ax bond angle in NpO
22+ is exactly 180° owing to its centrosymmetric structure in this compound. These structural data are common to those observed in NpO
2(NO
3)
2(OPPh
3)
2 (Np≡O
ax: 1.739(10) Å, Np−O
NO3: 2.53 Å (mean), Np−O
OPPh3: 2.363(8) Å), which is a
trans-dinitrato NpO
22+ complex with two monodentate ligands solely known so far [
19]. There are several related structures in which the equatorial coordination is constrained in a
cis-geometry by additional bidentate ligands, such as 2,2′-bipyridyl (bpy) and BrCH
2COO
−, or by dimerization through two μ-OH
− bridges [
20,
21,
22]. Regardless of such
cis-
trans isomerization, the Np≡O
ax distance of the current compound (1.739(10) Å) is not very different from the reported
cis-structures (1.73–1.76 Å). In contrast, it seems to be difficult to directly compare the Np−O
NO3 interactions in [NpO
2(NO
3)
2(
L1)]
n in the
trans-geometry (mean: 2.53 Å) to those in the known
cis-complexes owing to the wider variety in Np−O
NO3 lengths of the latter series from 2.47 Å to 2.56 Å.
As shown in
Table 2, [NpO
2(NO
3)
2(
L1)]
n is isostructural with [UO
2(NO
3)
2(
L1)]
n in terms of molecular structure as well, where the contribution of the actinide contraction is not clearly observed. Charushnikova et al. reported a molecular structure of [N(CH
3)
4][Np
VO
2(NO
3)
2(H
2O)
2], where Np is pentavalent [
23]. Although the coordination geometry around its Np
5+ center was also hexagonal bipyramidal in a similar manner to the current compound, the bond distances of this Np(V) complex (Np≡O
ax: 1.823(1) Å, Np−O
NO3: 2.62 Å (mean), Np−O
H2O: 2.473(1) Å) are critically different from those in
Table 2. The significantly shorter interactions around the Np center of [NpO
2(NO
3)
2(
L1)]
n reported here indicate that its hexavalence is maintained.
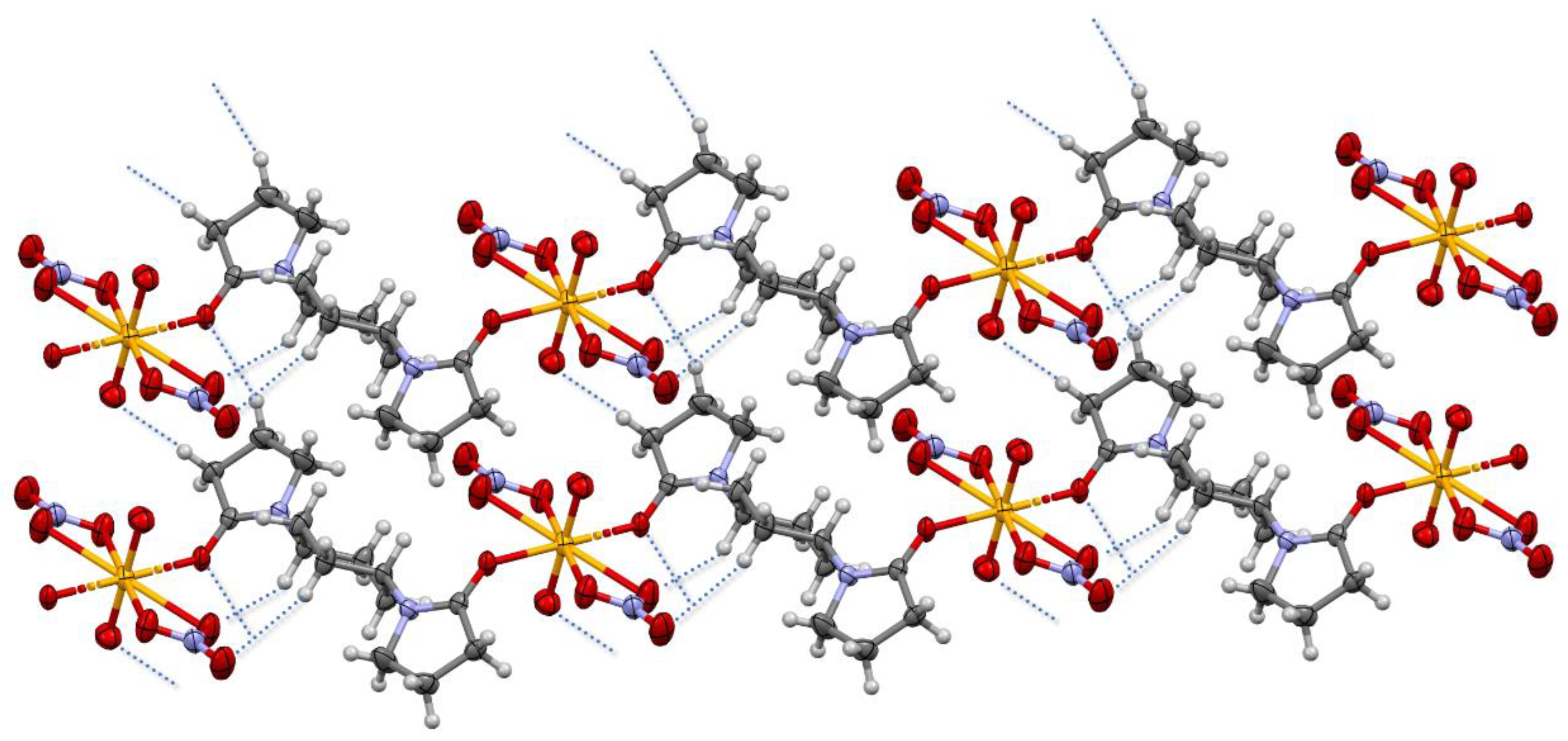
Each crystal lattice of this compound contains a [NpO
2(NO
3)
2(
L1)] monomer, which polymerizes along the
c-axis (
Figure 3,
Figure 4 and
Figure S1). In the directions of
a- and
b-axes, the [NpO
2(NO
3)
2(
L1)]
n coordination polymers stack each other through C−H···O interactions between the independent 1D chains (
D⋯
A: C⋯O
ax = 3.46–3.55 Å, C⋯O
NO3 = 3.28–3.63 Å, C⋯O
L = 3.61–3.65 Å;
D−H⋯
A: C−H⋯O
ax = 2.58–2.60 Å, C−H ⋯O
NO3 = 2.42–2.70 Å, C−H⋯O
L = 2.68–2.69 Å). These packing trends are also common to those of [UO
2(NO
3)
2(
L1)]
n [
9].
Previously, we defined a mean volume of one C atom of an alkyl group (R,
Figure 1) of NRPs as a compactness parameter (
CP, Å
3) as follows [
12].
where
V and
Z are the cell volume and the number of UO
2(NO
3)
2(NRP)
2 units in a crystal lattice, respectively. The subscript “0” indicates those of UO
2(NO
3)
2(2-pyrrolidone)
2, where NRP with R = H (
Figure 1) is employed.
NC is the number of C atoms in R.
CP can be compared as a measure of packing efficiency of UO
2(NO
3)
2(NRP)
2, and is an important factor to govern the solubility of UO
22+ in HNO
3(aq), i.e., recovery yield of U in our NUMAP reprocessing for the nuclear fuel recycling. Thereafter, this concept was expanded to the coordination polymers of [UO
2(NO
3)
2(DHNRP)]
n, where half of the number of C atoms in R’ of DHNRP (
Figure 1) was taken as
NC owing to its stoichiometric ratio [
2,
9]. Based on the structural similarity with the UO
22+ analogue discussed above,
CP of [NpO
2(NO
3)
2(
L1)]
n was calculated to be 16.4 Å
3 (
V0/
Z0 = 383.9 Å
3 at 293 K) [
9]. This is slightly greater than that of [UO
2(NO
3)
2(
L1)]
n (
CP = 15.6 Å
3), but still much smaller than those of UO
2(NO
3)
2(NRP)
2 complexes (
CP = 20.4 − 30.3 Å
3). Therefore, the higher packing efficiency of NpO
2(NO
3)
2 units is still maintained in [NpO
2(NO
3)
2(
L1)]
n.
Up to now, we have not found any significant differences arising from variation in the hexavalent actinide center of [AnO
2(NO
3)
2(
L1)]
n from the viewpoint of structural chemistry. Seemingly, such a similarity might allow us to anticipate co-crystallization of any AnO
22+ ions from HNO
3(aq) after loading
L1 to form the heterometallic 1D coordination polymer. Nevertheless, we should reject this expectation, because the recovery yield of NpO
22+ from 3 M HNO
3(aq) was only 44% after loading an equimolar amount of
L1 at 293 K, as described above. This means that the solubility of [NpO
2(NO
3)
2(
L1)]
n is 16 mM under this condition, which is one order of magnitude higher than that of [UO
2(NO
3)
2(
L1)]
n (2.48 mM) [
9]. It is rather close to that of UO
2(NO
3)
2(NRP)
2 (R = cyclohyexyl, 17.6 mM,
CP = 21.4 Å
3) [
2]. Here, we found a uniqueness of NpO
22+.
As mentioned above, the structural features cannot explain this gap. We wonder whether the difference in solubility of [AnO
2(NO
3)
2(
L1)]
n between An = U and Np could be ascribed to the thermodynamic stability of the complexation around AnO
22+. Indeed, stability constants (log
β) of AnO
22+ complexes in several ligand systems (CO
32−, CH
3COO
−, F
−) sequentially decrease with an increase in the atomic number of the center metal, as summarized in
Table S1 [
16,
24]. We are also aware that such a trend is not always clear for other weaker ligands (Cl
−, SO
42−), and that log
β of NO
3− is only available for UO
22+ despite the highest relevance to the current work. Although it is still difficult to know exactly why the solubility of [NpO
2(NO
3)
2(
L1)]
n is unexpectedly higher than the UO
22+ analogue, this knowledge provides an important implication to consider the recovery of PuO
22+ using
L1 from HNO
3(aq) in our NUMAP reprocessing for nuclear fuel recycling [
2].
Using
rac-
L2 (see
Figure 1), we previously confirmed that [UO
2(NO
3)
2(
rac-
L2)]
n deposited from 3 M HNO
3(aq) [
9]. Therefore, we examined what happens in a mixture of NpO
22+ with
rac-
L2 as follows. The aqueous solution with 3 M HNO
3 and 100 mM NpO
22+ (25 μL) was placed at the bottom of the
ϕ5 mm glass tube, followed by subsequent loading of a blank 3 M HNO
3(aq) (50 μL) and that dissolving 200 mM
rac-
L2 (12.5 μL), where NpO
22+:
rac-
L2 = 1:1. Finally, nothing deposited even after several days passed. However, this result is not very surprising, because we have already known that
rac-
L2 affords a more soluble coordination polymer of [UO
2(NO
3)
2(
rac-
L2)]
n (14.8 mM) compared with [UO
2(NO
3)
2(
L1)]
n (2.48 mM) [
9]. Accordingly, the solubility of the NpO
22+ analogue with
rac-
L2 would also be higher than that of
L1 described above (16 mM). When the sample solution was fully mixed, the total concentration of NpO
22+ was 28.6 mM. While there are no reasons to exclude the possibility of formation of the [NpO
2(NO
3)
2(
rac-
L2)]
n coordination polymer in a similar manner to [UO
2(NO
3)
2(
rac-
L2)]
n, its solubility in 3 M HNO
3(aq) would be higher than 28.6 mM, the total concentration of NpO
22+ loaded to the reaction mixture of the current experimental runs.
4. Experimental
4.1. Materials
Caution! 237Np is a radioactive isotope emitting alpha radiation (specific activity: 2.63×107 Bq g–1 with T1/2 = 2.14 × 106 years). It has to be handled in dedicated facilities with appropriate equipment for radioactive materials to avoid health risks caused by radiation exposure. All of the operations in handling Np were performed in a dedicated glove box in the control area of HZDR. Furthermore, HNO3 used in this work should be used under great caution for chemical safety because of its strong acidity and oxidizing nature.
All reagents used were of reagent grade and used as received, if not specified. The
L1 and
rac-
L2 ligands were synthesized as described below [
9]. Neptunyl(VI) nitrate hydrate (Np
VIO
2(NO
3)
2·
nH
2O,
n ~ 5) was prepared by dissolving NpO
2 (29.8 mg, 0.111 mmol) in conc. HNO
3 (aq.). This solution was refluxed for two days and concentrated to near to dryness on a hot plate. The final residue of Np
VIO
2(NO
3)
2·
nH
2O was dissolved in 1.11 mL of 3 M HNO
3 (aq.) to prepare 100 mM Np
VIO
2(NO
3)
2 with 3 M HNO
3 stock solution. The concentration of NpO
22+ was determined by the γ-ray spectrometry.
4.2. Synthesis of L1
In a 200 mL round bottom flask, trans-1,4-cyclohexanediamine (1.225 g, 10.73 mmol, TCI), K2CO3 (6.10 g, 44.2 mmol, Wako), and THF (80 mL, Wako) were loaded. Under cooling on an ice bath and vigorous stirring, 4-chlorobutyryl chloride (2.45 mL, 3.09 g, 21.90 mmol, TCI) was slowly added through a dropping funnel. This mixture was stirred at 0 °C for 2 h, and then further reacted at room temperature overnight. After removal of the solvent by evaporation under reduced pressure, water (70 mL) was loaded to the residue, followed by sonication and stirring at room temperature. The colorless insoluble solid was recovered by filtration, washed with H2O several times, and dried under vacuum to obtain N,N’-trans-cyclohexane-1,4-diylbis(4-chlorobutanamide) (2.50 g, 72% yield). 1H NMR (δ/ppm vs. TMS, CDCl3, 399.78 MHz) 5.47 (NH, bs, 2H), 3.76 (N−CHax, br, 2H), 3.58 (CH2Cl, t, 4H), 2.33 (CH2, t, 4H), 2.11 (CH2, quintet, 4H), 2.01 (CHeq, d, 4H), 1.27 (CHax, td, 4H). 13C NMR (δ/ppm vs. TMS, CDCl3, 100.53 MHz) 171.03, 47.71, 44.67, 33.50, 31.79, 28.22.
This precursor (2.42 g, 7.44 mmol) was loaded in a 200 mL round bottom flask together with CH2Cl2 (70 mL), followed by cooling on the ice bath. To this solution, potassium tert-butoxide (1.85 g, 16.5 mmol) dissolved in THF (40 mL) was slowly added through a dropping funnel. The reaction mixture was stirred at 0 °C overnight. After passing through a Celite pad, the filtrate was concentrated by a rotary evaporator. Adding hexane (40 mL) to the residual oil under sonication allowed to form a colorless solid, which was recovered by suction filtration and dried under vacuum to obtain L1 (1.54 g) in 83% yield. 1H NMR (δ/ppm vs. TMS, CDCl3, 399.78 MHz) 3.97 (N−CHax, br, 2H), 3.33 (CH2, t, 4H), 2.39 (CH2, t, 4H), 2.00 (CH2, quintet, 4H), 1.77 (CHeq, d, 4H), 1.60 (CHax, td, 4H). 13C NMR (δ/ppm vs. TMS, CDCl3, 100.53 MHz) 171.03, 47.71, 44.67, 33.50, 31.79, 28.22.
4.3. Synthesis of rac-L2
In a 500 mL round bottom flask, trans-1,2-cyclohexanediamine (5.08 g, 44.4 mmol, TCI), triethylamine (8.99 g, 88.8 mmol, Kanto), and THF (200 mL, Wako) were loaded. Under cooling on an ice bath and vigorous stirring, 4-chlorobutyryl chloride (9.95 mL, 12.5 g, 88.9 mmol, TCI) was slowly added through a dropping funnel. This mixture was stirred at 0 °C for 2 h, and then further reacted at room temperature overnight. After filtration of the reaction mixture, the obtained solid was dispersed in H2O (150 mL). The insoluble colorless solid was recovered by filtration, washed with H2O several times, and dried under vacuum to obtain N,N’-trans-cyclohexane-1,2-diylbis(4-chlorobutanamide) (6.58 g, 46% yield). The filtrate after the reaction was concentrated by the rotary evaporator. To the residue, CH2Cl2 (130 mL) and 2 M HCl aq (10 mL) were loaded, followed by vigorous stirring of this mixture. The organic layer was separated from the aqueous phase and mixed with K2CO3 and MgSO4. The separated supernatant was concentrated and triturated with cold hexane (100 mL) in the ice bath. The deposited colorless solid was recovered by filtration, washed with cold hexane, and dried under vacuum to additionally obtain N,N’-trans-cyclohexane-1,2-diylbis(4-chlorobutanamide) (3.54 g, 25% yield). 1H NMR revealed that both compounds prepared here were identical to each other. The total yield was 71%. 1H NMR (δ/ppm vs. TMS, CDCl3, 399.78 MHz) 5.58 (NH, bd, 2H), 3.94 (N−CHax, br, 2H), 3.62 (CH2Cl, t, 4H), 2.36 (CH2, t, 4H), 2.12 (CH2, quintet, 4H), 1.77 (CHeq, m, 4H), 1.57 (CHax, m, 4H). 13C NMR (δ/ppm vs. TMS, CDCl3, 100.53 MHz) 170.98, 45.78, 44.69, 33.48, 28.37, 28.16.
This precursor (3.54 g, 11.0 mmol) was loaded in a 200 mL round bottom flask together with THF (110 mL), followed by cooling in the ice bath. To this solution, potassium tert-butoxide (2.57 g, 22.9 mmol) dissolved in THF (30 mL) was slowly added through a dropping funnel. The reaction mixture was stirred at 0 °C for 1 h and further reacted at room temperature for 6 h. After passing through a Celite pad, the filtrate was concentrated by a rotary evaporator. The residue was mixed with CH2Cl2 (80 mL) and 1 M HCl aq (10 mL), followed by stirring for several minutes. The separated organic layer was mixed with K2CO3 and MgSO4. After removal of the solid residues, the filtrate was evaporated under reduced pressure to dryness to obtain a colorless microcrystalline compound of rac-L2 (2.46 g) at a 90% yield. 1H NMR (δ/ppm vs. TMS, CDCl3, 399.78 MHz) 4.04 (N−CHax, br, 2H), 3.51 (CH2, t, 4H), 2.39 (CH2, t, 4H), 2.03 (CH2, quintet, 4H), 1.87 (CHeq, m, 4H), 1.730 (CHax, m, 4H). 13C NMR (δ/ppm vs. TMS, CDCl3, 100.53 MHz) 175.02, 48.02, 45.73, 31.42, 27.18, 18.42.
4.4. Synthesis of [NpVIO2(NO3)2(L1)]n
A stock solution of 100 mM NpVIO22+ in 3 M HNO3 (25 μL), a blank 3 M HNO3(aq) (50 μL), and a stock solution of 200 mM L1 in 3 M HNO3(aq) were layered from bottom to top of a glass tube (ca. ϕ5 mm diameter) in a stepwise manner, followed by standing in a silent place in the glovebox at 293 K. After one day passed, brown block crystals of [NpVIO2(NO3)2(L1)]n were obtained at 44% yield, which was estimated by the following procedure. The supernatant (1 μL) was loaded into a ca. ϕ10 mm glass vial and subjected to γ-ray spectrometry, performed at VKTA Dresden. The γ-ray emission from the sample was counted for 25 min real time plus 0.71% dead time.
4.5. Crystallographic Analysis
The X-ray diffraction data of the well-shaped single crystal of [Np
VIO
2(NO
3)
2(
L1)]
n were collected by the Bruker D8 VENTURE diffractometer equipped with hybrid pixel array detector and graphite monochromated Mo
Kα radiation (
λ = 0.71073 Å). A selected single crystal of the sample was mounted on a MiTeGen Dual Thickness MicroMounts with mineral oil. Intensity data were collected by taking oscillation photographs. Reflection data were integrated with the Bruker SAINT software package. Absorption correction was applied using the strong absorber option of SADAS22. The structures were solved by the direct method and refined anisotropically for non-hydrogen atoms by full-matrix least-squares calculations with the SHELX program suite [
27]. Each refinement was continued until all shifts were smaller than one-third of the standard deviations of the parameters involved. Hydrogen atoms were located at the calculated positions. All hydrogen atoms were constrained to an ideal geometry with C–H = 0.95 Å. The thermal parameters of all hydrogen atoms were related to those of their parent atoms by
U(H) = 1.2
Ueq(C). All calculations were performed using the
Olex2 crystallographic software program package [
28]. The crystallographic data of [NpO
2(NO
3)
2(
L1)]
n are summarized in
Table 1 together with those of [UO
2(NO
3)
2(
L1)]
n, which we reported previously [
9].









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}