
The Critical Role of Ligand Flexibility on the Activity of Free and Immobilized Mn Superoxide Dismutase Mimics



 and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of Ligands, Complexes and Hybrid Materials
2.1.1. Synthesis of 1,3-Bis[(2-Pyridilmethyl)(Propargyl)Amino]Propane (Pypapn)
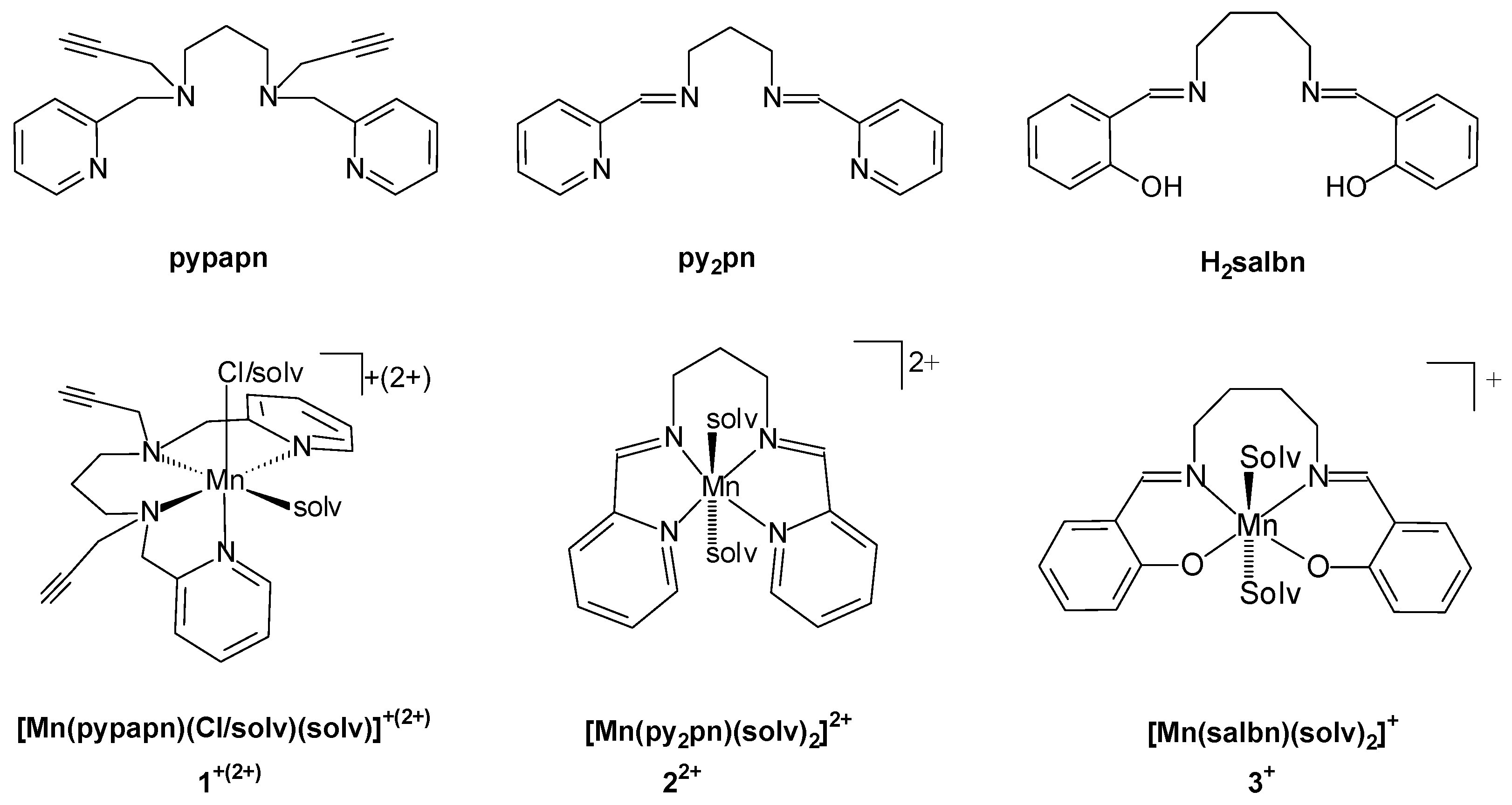
2.1.2. Synthesis of [Mn(Pypapn)Cl]Cl⋅0.5 H2O (1)
2.1.3. Synthesis of [Mn2(Py2pn)3(ClO4)2](ClO4)2⋅2H2O (2)
2.1.4. Synthesis of [Mn2(Salbn)2OH]ClO4 (3)
2.1.5. Synthesis of “Click” Modified Silica Pypntriazole@OP-MS
2.1.6. Encapsulation of Complexes 2 and 3 in Mesoporous Silica SBA-15
2.2. Analytical and Physical Measurements
2.2.1. Analytical Measurements
2.2.2. Spectroscopy Measurements
2.2.3. Conductivity and Electrochemical Measurements
2.2.4. Electron Microscopy Measurements
2.2.5. N2 Adsorption-Desorption Measurements
2.3. Indirect SOD Assay
2.4. Preparation of Potassium Superoxide Solutions
3. Results and Discussion
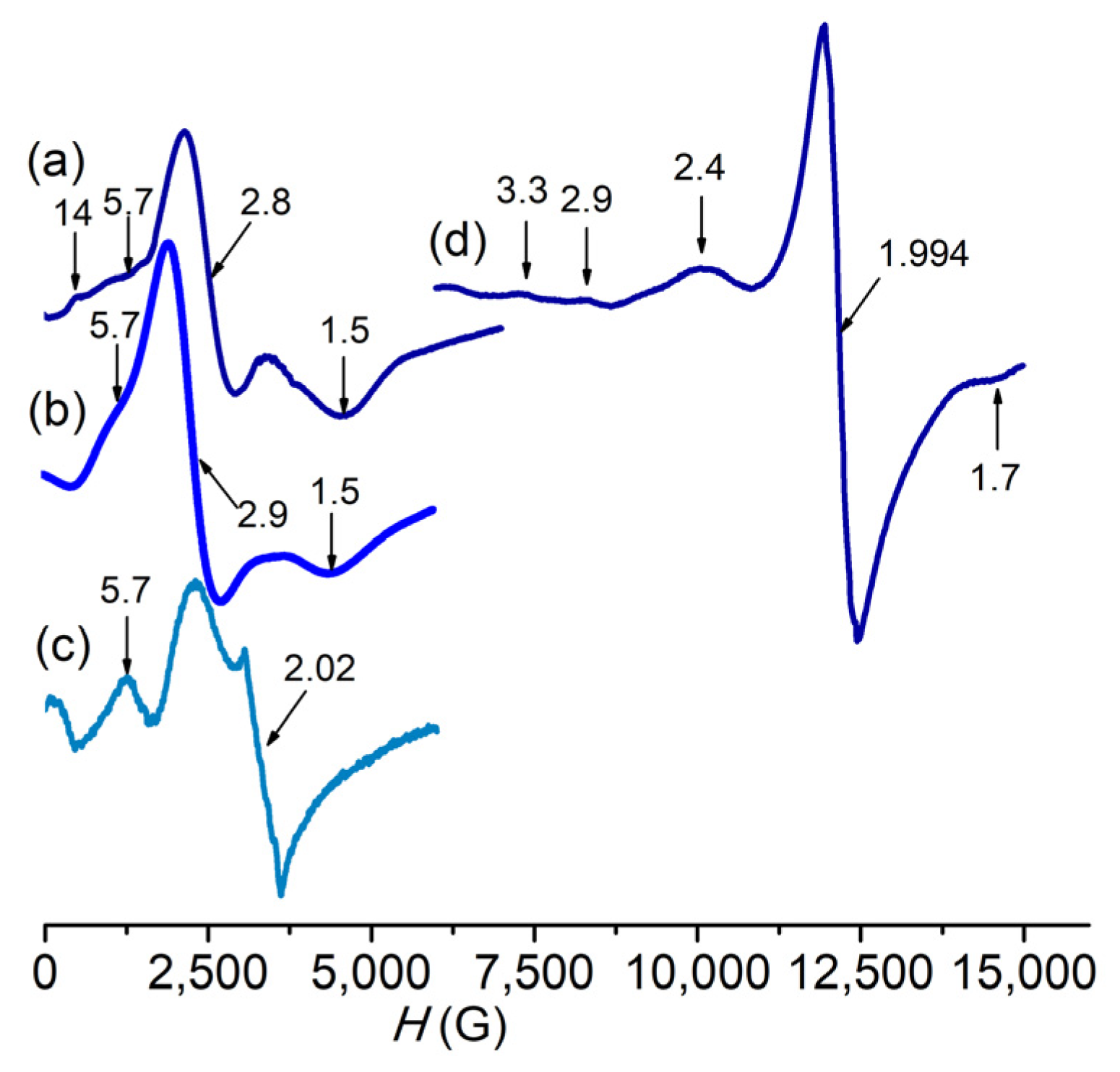
3.1. Characterization of Complexes
3.2. Electrochemical Studies
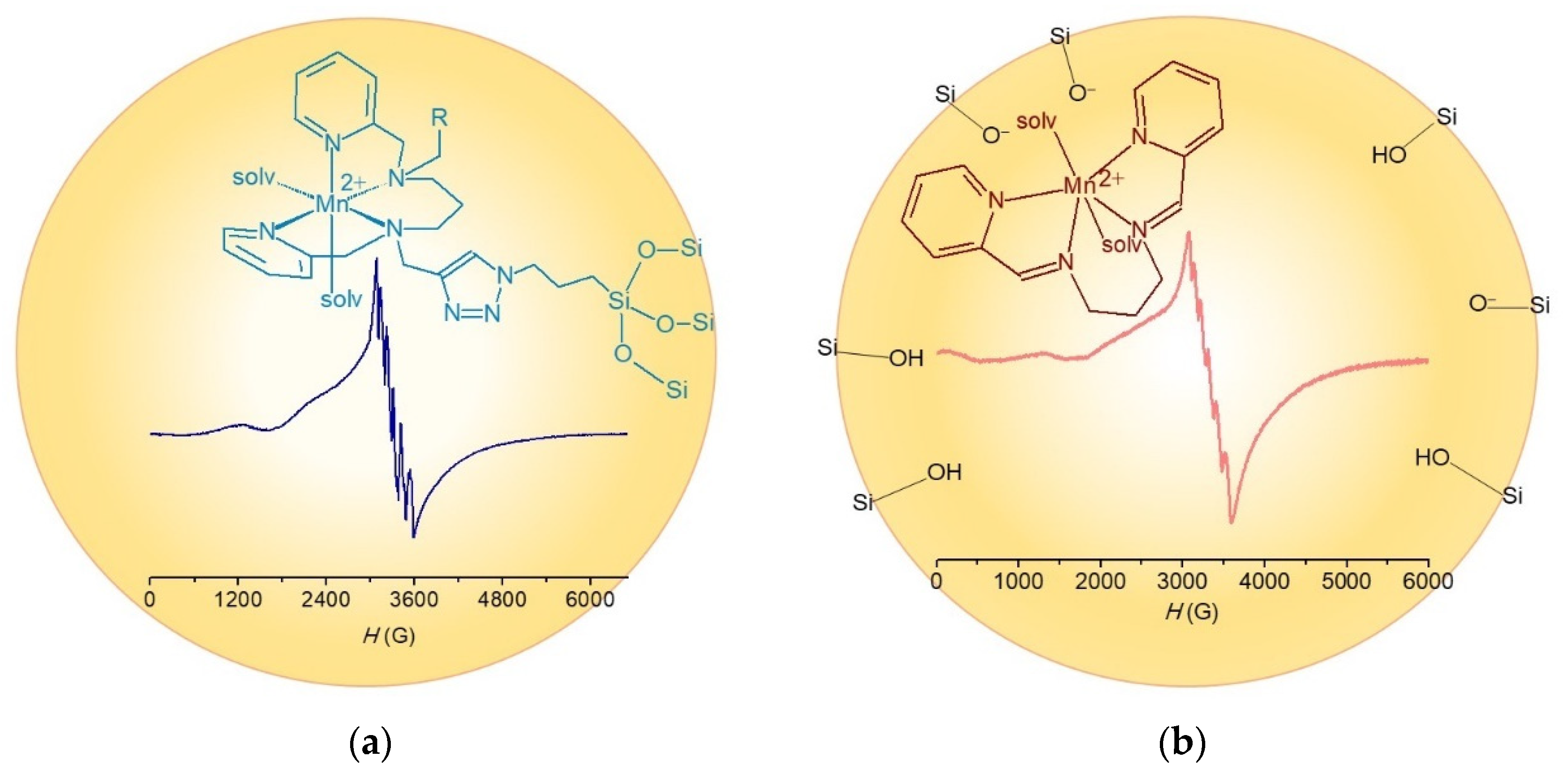
3.3. Synthesis and Characterization of Modified Mesoporous Silicas
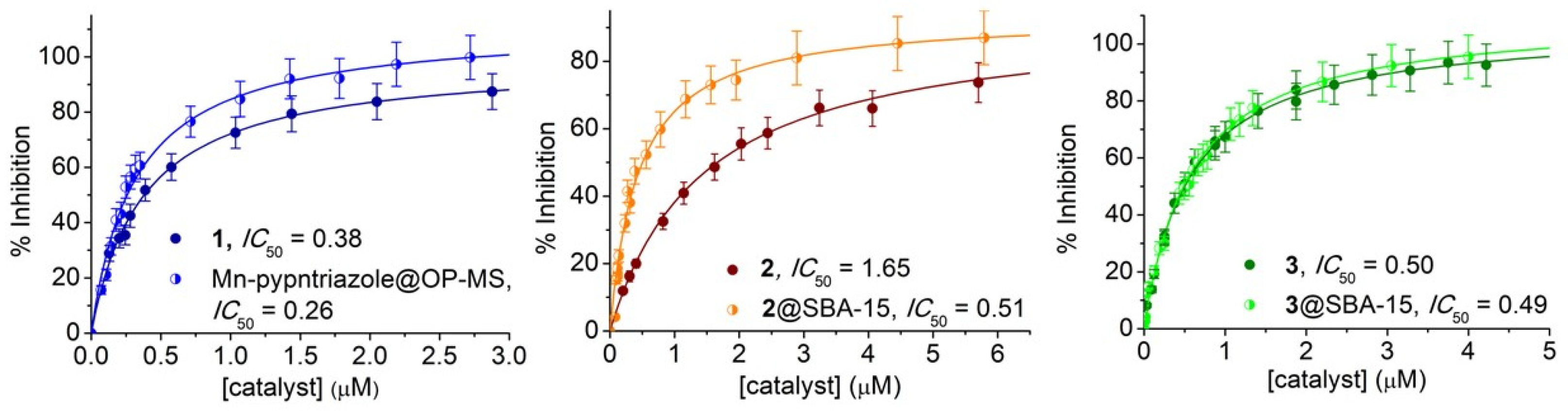
3.4. SOD Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Ligand Donor Sites | 106 kMcF (M−1s−1) | E1/2 (II/III) (V vs. SCE) | Ref. |
|---|---|---|---|---|---|
| 1 | 12+a | N4 | 5.97 | 1.1 (Epa) | This work |
| 2 | [Mn(II)(NTB)(terphthalate)] | N4 | 4.3 | 0.059 | [69] |
| 3 | 22+ | N4 | 1.40 | >1 | This work |
| 4 | [Mn(III)(bpb)Cl(H2O)] | N4 | 0.93 | −0.017 | [70] |
| 5 | [Mn(II)(PClNOL)Cl2] | N3O | 9.4 | 0.806 | [71] |
| 6 | [Mn(II)(PBMPA)Cl(H2O)] | N3O | 4.9 | 0.47 | [72] |
| 7 | [Mn(II)(BMPG)(H2O)]+ | N3O | 4.8 | 0.44 (Epa) | [73] |
| 8 | [Mn(II)(BIG)(H2O)2]+ | N3O | 1.5 | 0.756 (Epa) | [73] |
| 9 | 3+ | N2O2 | 4.54 | 0.055 | This work |
| 10 | [Mn(III)(hbpapn)(H2O)2]+ | N2O2 | 3.90 | 0.42 (III/IV) | [19] |
| 11 | [Mn(III)(pyr2pn)(H2O)2]+ | N2O2 | 1.84 | ND | [74] |
| 12 | [Mn(III)(salpn)(H2O)2]+ | N2O2 | 1.53 | −0.14 | [58] |
| 13 | [Mn(III)(X-salen)(H2O)2]+ | N2O2 | 0.6 | −0.237 to 0.031 | [14,75] |
| 14 | MnSOD | N3O | 800 | 0.049 | [2,78] |
| Immobilized Catalyst | 106 kMcF (M−1 s−1) | ||||
| 15 | Mn-pypntriazole@OP-MS | 8.73 | |||
| 16 | 2@SBA-15 | 4.45 | |||
| 17 | 3@SBA-15 | 4.63 |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, B.; Chen, Y.; Shi, J. Reactive Oxygen Species (ROS)-Based Nanomedicine. Chem. Rev. 2019, 119, 4881–4985. [Google Scholar]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar]
- Dixon, M.M.; Pattridge, K.A.; Stallings, W.C.; Fee, J.A.; Ludwig, M.L. Structure—Function in Escherichia coli Iron Superoxide Dismutase: Comparisons with the Manganese Enzyme from Thermus thermophilus. Biochemistry 1995, 34, 1646–1660. [Google Scholar]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative Stress and Neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar]
- Salvemini, D.; Muscoli, C.; Riley, D.P.; Cuzzocrea, S. Superoxide Dismutase Mimetics. Pulm. Pharmacol. Ther. 2002, 15, 439–447. [Google Scholar]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St. Clair, D.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar]
- Bonetta, R. Potential Therapeutic Applications of MnSODs and SOD-Mimetics. Chem. Eur. J. 2018, 24, 5032–5041. [Google Scholar] [CrossRef]
- Policar, C.; Bouvet, J.; Bertrand, H.C.; Delsuc, N. SOD mimics: From the toolbox of the chemists to cellular studies. Curr. Opin. Chem. Biol. 2022, 67, 102109. [Google Scholar] [CrossRef]
- Signorella, S.; Palopoli, C.; Ledesma, G. Rationally designed mimics of antioxidant manganoenzymes: Role of structural features in the quest for catalysts with catalase and superoxide dismutase activity. Coord. Chem. Rev. 2018, 365, 75–102. [Google Scholar]
- Schanne, G.; Zoumpoulaki, M.; Gazzah, G.; Vincent, A.; Preud’Homme, H.; Lobinski, R.; Demignot, S.; Seksik, P.; Delsuc, N.; Policar, C. Inertness of Superoxide Dismutase Mimics Mn(II) Complexes Based on an Open-Chain Ligand, Bioactivity, and Detection in Intestinal Epithelial Cells. Oxidative Med. Cell. Longev. 2022, 2022, 3858122. [Google Scholar]
- Garda, Z.; Molnár, E.; Hamon, N.; Barriada, J.L.; Gómez, D.E.; Váradi, B.; Nagy, V.; Pota, K.; Kálmán, F.K.; Tóth, I.; et al. Complexation of Mn(II) by Rigid Pyclen Diacetates: Equilibrium, Kinetic, Relaxometric, Density Functional Theory, and Superoxide Dismutase Activity Studies. Inorg. Chem. 2021, 60, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.; Bernard, A.-S.; Delsuc, N.; Quévrain, E.; Gazzah, G.; Lai, B.; Chain, F.; Langella, P.; Bachelet, M.; Masliah, J.; et al. A Cell-Penetrant Manganese Superoxide Dismutase (MnSOD) Mimic Is Able To Complement MnSOD and Exerts an Antiinflammatory Effect on Cellular and Animal Models of Inflammatory Bowel Diseases. Inorg. Chem. 2017, 56, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Kálmán, F.K.; Tircsó, G. Kinetic Inertness of the Mn2+ Complexes Formed with AAZTA and Some Open-Chain EDTA Derivatives. Inorg. Chem. 2012, 51, 10065–10067. [Google Scholar] [CrossRef]
- González-Riopedre, G.; Fernández-García, M.; Gómez-Fórneas, E.; Maneiro, M. Biomimetic Catalysts for Oxidation of Veratryl Alcohol, a Lignin Model Compound. Catalysts 2013, 3, 232–246. [Google Scholar]
- Mureseanu, M.; Filip, M.; Bleotu, I.; Spinu, C.I.; Marin, A.H.; Matei, I.; Parvulescu, V. Cu(II) and Mn(II) Anchored on Functionalized Mesoporous Silica with Schiff Bases: Effects of Supports and Metal–Ligand Interactions on Catalytic Activity. Nanomaterials 2023, 13, 1884. [Google Scholar] [CrossRef] [PubMed]
- Rana, B.S.; Jain, S.L.; Singh, B.; Bhaumik, A.; Sain, B.; Sinha, A.K. Click on silica: Systematic immobilization of Co(II)Schiff bases to the mesoporous silicavia click reaction and their catalytic activity for aerobic oxidation of alcohols. Dalton Trans. 2010, 39, 7760–7767. [Google Scholar] [CrossRef]
- Chaignon, J.; Stiriba, S.-E.; Lloret, F.; Yuste, C.; Pilet, G.; Bonneviot, L.; Albela, B.; Castro, I. Bioinspired manganese(II) complexes with a clickable ligand for immobilisation on a solid support. Dalton Trans. 2014, 43, 9704–9713. [Google Scholar] [CrossRef]
- Nodzewska, A.; Wadolowska, A.; Watkinson, M. Recent advances in the catalytic oxidation of alkene and alkane substrates using immobilized manganese complexes with nitrogen containing ligands. Coord. Chem. Rev. 2019, 382, 181–216. [Google Scholar]
- Richezzi, M.; Palopoli, C.; Pellegri, N.; Hureau, C.; Signorella, S.R. Synthesis, characterization and superoxide dismutase activity of a biomimetic Mn(III) complex covalently anchored to mesoporous silica. J. Inorg. Biochem. 2022, 237, 112026. [Google Scholar] [CrossRef]
- Liberman, A.; Mendez, N.; Trogler, W.C.; Kummel, A.C. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. [Google Scholar]
- Sun, R.; Qiao, P.; Wang, Z.; Wang, W. Monodispersed large-mesopore mesoporous silica nanoparticles enabled by sulfuric acid assisted hydrothermal process. Microporous Mesoporous Mater. 2021, 317, 111023. [Google Scholar] [CrossRef]
- Ebralidze, I.I.; Leitus, G.; Shimon, L.J.W.; Wang, Y.; Shaik, S.; Neumann, R. Structural variability in manganese(II) complexes of N,N′-bis(2-pyridinylmethylene) ethane (and propane) diamine ligands. Inorg. Chim. Acta 2009, 362, 4713–4720. [Google Scholar] [CrossRef]
- Kadwa, E.; Bala, M.D.; Friedrich, H.B. Characterisation and application of montmorillonite-supported Fe Schiff base complexes as catalysts for the oxidation of n-octane. Appl. Clay Sci. 2014, 95, 340–347. [Google Scholar] [CrossRef]
- Thielemann, J.P.; Girgsdies, F.; Schlögl, R.; Hess, C. Pore structure and surface area of silica SBA-15: Influence of washing and scale-up. Beilstein J. Nanotechnol. 2011, 2, 110–118. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Villarroel Rocha, J.; Barrera, D.; Sapag, K. Improvement in the Pore Size Distribution for Ordered Mesoporous Materials with Cylindrical and Spherical Pores Using the Kelvin Equation. Top. Catal. 2011, 54, 121–134. [Google Scholar] [CrossRef]
- Beauchamps, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Liao, Z.-R.; Zheng, X.-F.; Luo, B.-S.; Shen, L.-R.; Li, D.-F.; Liu, H.-L.; Zhao, W. SOD-like activities of manganese-containing complexes with N,N,N,N-tetrakis(2-benzimidazolyl methyl)-1,2-ethanediamine (EDTB). Polyhedron 2001, 20, 2813–2821. [Google Scholar] [CrossRef]
- Hyland, K.; Auclair, C. The formation of superoxide radical anions by a reaction between O2, OH− and dimethyl sulfoxide. Biochem. Biophys. Res. Commun. 1981, 102, 531–537. [Google Scholar] [CrossRef]
- Dowsing, R.D.; Nieuwenhuijse, B.; Reedijk, J. Pyrazoles and imidazoles as ligands. X. electron paramagnetic resonance spectra of MnII in a tetragonal environment of four pyrazoles and two anions. Inorg. Chim. Acta 1971, 5, 301–304. [Google Scholar] [CrossRef]
- Warzeska, S.T.; Miccichè, F.; Mimmi, M.C.; Bouwman, E.; Kooijman, H.; Spek, A.L.; Reedijk, J. Tuning the coordination mode in mononuclear manganese complexes by changing the steric bulk of the carboxylates. J. Chem. Soc. Dalton Trans. 2001, 3507–3512. [Google Scholar] [CrossRef]
- Hureau, C.; Blondin, G.; Charlot, M.-F.; Philouze, C.; Nierlich, M.; Césario, M.; Anxolabéhère-Mallart, E. Synthesis, Structure, and Characterization of New Mononuclear Mn(II) Complexes. Electrochemical Conversion into New Oxo-Bridged Mn2(III,IV) Complexes. Role of Chloride Ions. Inorg. Chem. 2005, 44, 3669–3683. [Google Scholar] [CrossRef] [PubMed]
- Duboc, C.; Collomb, M.-N.; Neese, F. Understanding the Zero-Field Splitting of Mononuclear Manganese(II) Complexes from Combined EPR Spectroscopy and Quantum Chemistry. Appl. Magn. Reson. 2010, 37, 229–245. [Google Scholar] [CrossRef]
- Jabłonska-Wawrzycka, A.; Barszcz, B.; Zienkiewicz, M.; Hodorowicz, M.; Jezierska, J.; Stadnicka, K.; Lechowicz, Ł.; Kaca, W. Eight- and six-coordinated Mn(II) complexes of heteroaromatic alcohol and aldehyde: Crystal structure, spectral, magnetic, thermal and antibacterial activity studies. Spectrochim. Acta Part A 2014, 129, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Duboc, C.; Phoeung, T.; Zein, S.; Pécaut, J.; Collomb, M.N.; Neese, F. Origin of the Zero-Field Splitting in Mononuclear Octahedral Dihalide MnII Complexes: An Investigation by Multifrequency High-Field Electron Paramagnetic Resonance and Density Functional Theory. Inorg. Chem. 2007, 46, 4905–4916. [Google Scholar] [CrossRef] [PubMed]
- Mantel, C.; Baffert, C.; Romero, I.; Deronzier, A.; Pécaut, J.; Collomb, M.-N.; Duboc, C. Structural Characterization and Electronic Properties Determination by High-Field and High-Frequency EPR of a Series of Five-Coordinated Mn(II) Complexes. Inorg. Chem. 2004, 43, 6455–6463. [Google Scholar] [CrossRef]
- Duboc, C. Determination and prediction of the magnetic anisotropy of Mn ions. Chem. Soc. Rev. 2016, 45, 5834–5847. [Google Scholar] [CrossRef]
- Bucher, C.; Duval, E.; Barbe, J.-M.; Verpeaux, J.-N.; Amatore, C.; Guilard, R.; Le Pape, L.; Latour, J.-M.; Dahaoui, S.; Lecomte, C. Synthesis, X-ray Structure, Electrochemical, and EPR Studies of a Pentacoordinated Mn(II) Tetramethylcyclam Complex. Inorg. Chem. 2001, 40, 5722–5726. [Google Scholar] [CrossRef]
- Hureau, C.; Groni, S.; Guillot, R.; Blondin, G.; Duboc, C.; Anxolabéhère-Mallart, E. Syntheses, X-ray Structures, Solid State High-Field Electron Paramagnetic Resonance, and Density-Functional Theory Investigations on Chloro and Aqua MnII Mononuclear Complexes with Amino-Pyridine Pentadentate Ligands. Inorg. Chem. 2008, 47, 9238–9247. [Google Scholar] [CrossRef]
- Ali, I.; Wani, W.A.; Saleem, K. Empirical Formulae to Molecular Structures of Metal Complexes by Molar Conductance. Synth. React. Inorg. Met.-Org. Chem. 2013, 43, 1162–1170. [Google Scholar] [CrossRef]
- Conradie, J.; Conradie, M.M.; Tawfiq, K.M.; Al-Jeboori, M.J.; Coles, S.J.; Wilson, C.; Potgieter, J.H. Novel dichloro(bis{2-[1-(4-methylphenyl)-1H-1,2,3-triazol-4-yl-κN3]pyridine-κN})metal(II) coordination compounds of seven transition metals (Mn, Fe, Co, Ni, Cu, Zn and Cd). Polyhedron 2018, 151, 243–254. [Google Scholar] [CrossRef]
- Farkas, D.; Hansson, Ö. An NMR study elucidating the binding of Mg(II) and Mn(II) to spinach plastocyanin. Regulation of the binding of plastocyanin to subunit PsaF of photosystem I. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Sultana, N.; Arayne, M.S.; Rizvi, S.B.S.; Haroon, U.; Mesaik, M.A. Synthesis, spectroscopic, and biological evaluation of some levofloxacin metal complexes. Med. Chem. Res. 2013, 22, 1371–1377. [Google Scholar] [CrossRef]
- Lavanant, H.; Virelizier, H.; Hoppilliard, Y. Reduction of Copper (II) Complexes by Electron Capture in an Electrospray Ionization Source. J. Am. Soc. Mass Spectrom. 1998, 9, 1217–1221. [Google Scholar] [CrossRef]
- Dikio, C.W.; Ejidike, I.P.; Mtunzi, F.M.; Klink, M.J.; Dikio, E.D. Hydrazide schiff bases of acetylacetonate metal complexes: Synthesis, spectroscopic and biological studies. Int. J. Pharm. Pharm. Sci. 2017, 9, 257–267. [Google Scholar] [CrossRef]
- Jambulingam, M.; Thangadurai, S.A.; Vijayabaskaran, M. Designing and Synthesis of Some Transition Metal Complexes Derived from Schiff Bases for Anti-Bacterial Activity. J. Med. Chem. Sci. 2022, 5, 10–18. [Google Scholar]
- Kanumfre, F.; de Lima, E.M.; Scheidt, G.; Carneiro, P.I.B.; Rosso, N.D. Potentiometric and spectrophotometric studies of MnII and NiII cimetidine complexes. J. Braz. Chem. Soc. 2010, 21, 800–805. [Google Scholar] [CrossRef]
- Biswas, S.; Mitra, K.; Adhikary, B. Studies on binuclear hydroxo/carboxylato-bridged manganese (III) complexes and a mononuclear manganese (III) complex involving salen type ligands. Transit. Met. Chem. 2005, 30, 586–592. [Google Scholar] [CrossRef]
- Zhou, H.-B.; Wang, H.-S.; Chen, Y.; Xu, Y.-L.; Song, X.-J.; Song, Y.; Zhang, Y.-Q.; You, X.Z. Synthesis, structure, magnetic properties and DFT calculations of two hydroxo-bridged complexes based on MnIII (Schiff-Bases). Dalton Trans. 2011, 40, 5999–6006. [Google Scholar] [CrossRef] [PubMed]
- Mundlapati, V.R.; Jena, P.; Acharya, A.N.; Kar, A.K.; Dash, A.C.; Biswal, H.S. Water exchange reaction of a manganese catalase mimic: Oxygen-17 NMR relaxometry study on (aqua)manganese(III) in a salen scaffold and its reactions in a mildly basic medium. RSC Adv. 2016, 6, 111739–111746. [Google Scholar] [CrossRef]
- Kurahashi, T. Reverse catalase reaction: Dioxygen activation via two-electron transfer from hydroxide to dioxygen mediated by a manganese(III) salen complex. Inorg. Chem. 2015, 54, 8356–8366. [Google Scholar] [CrossRef] [PubMed]
- Si, S.F.; Tang, J.K.; Liao, D.Z.; Jiang, Z.H.; Yan, S.P. Synthesis and crystal structure of oxo-bridged dimanganese(III) complex [MnIII(salpn)OH]2(CH3OH)(CH3CN)2. J. Mol. Struct. 2002, 606, 87–90. [Google Scholar] [CrossRef]
- Chu, H.-A.; Hillier, W.; Law, N.A.; Babcock, G.T. Vibrational spectroscopy of the oxygen-evolving complex and of manganese model compounds. Biochim. Biophys. Acta 2001, 1503, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Bonadies, J.A.; Maroney, M.J.; Pecoraro, V.L. Structurally diverse manganese(III) Schiff base complexes: Solution speciation via paramagnetic proton NMR spectroscopy and electrochemistry. Inorg. Chem. 1989, 28, 2044–2051. [Google Scholar] [CrossRef]
- Larson, E.J.; Pecoraro, V.L. The peroxide-dependent. μ2-O bond formation of manganese complex [MnIV SALPN(O)]2. J. Am. Chem. Soc. 1991, 113, 3810–3818. [Google Scholar] [CrossRef]
- Hoogenraad, M.; Ramkisoensing, K.; Driessen, W.L.; Kooijman, H.; Spek, A.L.; Bouwman, E.; Haasnoot, J.G.; Reedijk, J. Catalytic and electrochemical properties of new manganese(III) compounds of 2-(2′-hydroxyphenyl)-oxazoline (Hphox or HClphox). Molecular structures of [Mn(Clphox)2(MeOH)2](ClO4) and [Mn(phox)2(MeOH)2][Mn(phox)2(ClO4)2](H2O)2. Inorg. Chim. Acta 2001, 320, 117–126. [Google Scholar] [CrossRef]
- Palopoli, C.; Gómez, G.; Foi, A.; Doctorovich, F.; Mallet-Ladeira, S.; Hureau, C.; Signorella, S. Dimerization, redox properties and antioxidant activity of two manganese (III) complexes of difluoro-and dichloro-substituted Schiff-base ligands. J. Inorg. Biochem. 2017, 167, 49–59. [Google Scholar] [CrossRef]
- Palopoli, C.; Ferreyra, J.; Conte-Daban, A.; Richezzi, M.; Foi, A.; Doctorovich, F.; Anxolabéhère-Mallart, E.; Hureau, C.; Signorella, S.R. Insights into Second-Sphere Effects on Redox Potentials, Spectroscopic Properties, and Superoxide Dismutase Activity of Manganese Complexes with Schiff-Base Ligands. ACS Omega 2019, 4, 48–57. [Google Scholar] [CrossRef]
- Torayama, H.; Nishide, T.; Asada, H.; Fujiwara, M.; Matsushita, T. Preparation and characterization of different two types of di-μ-oxo dimanganese (IV) complexes with tetradentate Schiff bases. Polyhedron 1998, 17, 105–118. [Google Scholar] [CrossRef]
- El Ghachtouli, S.; Ching, H.Y.V.; Lassalle-Kaiser, B.; Guillot, R.; Leto, D.F.; Chattopadhyay, S.; Jackson, T.A.; Dorlet, P.; Anxolabéhère-Mallart, E. Electrochemical formation of Mn III-peroxo complexes supported by pentadentate amino pyridine and imidazole ligands. Chem. Commun. 2013, 49, 5696–5698. [Google Scholar] [CrossRef]
- Singh, O.; Maji, A.; Singh, A.; Singh, N.; Ghosh, K. A new family of complexes derived from bis (imino) pyridine-type ligands: Crystal structures and bio-molecular interaction studies. Appl. Organomet. Chem. 2021, 35, e6177. [Google Scholar] [CrossRef]
- Rich, J.; Manrique, E.; Molton, F.; Duboc, C.; Collomb, M.-N.; Rodríguez, M.; Romero, I. Catalytic Activity of Chloro and Triflate Manganese(II) Complexes in Epoxidation Reactions: Reusable Catalytic Systems for Alkene Epoxidation. Eur. J. Inorg. Chem. 2014, 2014, 2663–2670. [Google Scholar] [CrossRef]
- Adhikary, J.; Chakraborty, A.; Dasgupta, S.; Chattopadhyay, S.K.; Kruszynski, R.; Trzesowska-Kruszynska, A.; Stepanović, S.; Gruden-Pavlović, M.; Swart, M.; Das, D. Unique mononuclear MnII complexes of an end-off compartmental Schiff base ligand: Experimental and theoretical studies on their bio-relevant catalytic promiscuity. Dalton Trans. 2016, 45, 12409–12422. [Google Scholar] [PubMed]
- Melville, J.N.; Bernhardt, P.V. Electrochemical Exploration of Active Cu-Based Atom Transfer Radical Polymerization Catalysis through Ligand Modification. Inorg. Chem. 2021, 60, 9709–9719. [Google Scholar] [CrossRef] [PubMed]
- Signorella, S.; Hureau, C. Bioinspired functional mimics of the manganese catalases. Coord. Chem. Rev. 2012, 256, 1229–1245. [Google Scholar] [CrossRef]
- Malvi, B.; Sarkar, B.R.; Pati, D.; Mathew, R.; Ajithkumarb, T.G.; Sen Gupta, S. “Clickable” SBA-15 mesoporous materials: Synthesis, characterization and their reaction with alkynes. J. Mater. Chem. 2009, 19, 1409–1416. [Google Scholar] [CrossRef]
- Mercier, L.; Pinnavaia, T.J. Direct Synthesis of Hybrid Organic−Inorganic Nanoporous Silica by a Neutral Amine Assembly Route: Structure−Function Control by Stoichiometric Incorporation of Organosiloxane Molecules. Chem. Mater. 2000, 12, 188–196. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the detemination of surface area and porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Xiang, D.F.; Tan, X.S.; Hang, Q.W.; Tang, W.X.; Wu, B.-M.; Mak, T.C.W. Crystal structure and properties of a new five-coordinate manganese superoxide dismutase mimic. Inorg. Chim. Acta 1998, 277, 21–25. [Google Scholar]
- Lin, J.; Tu, C.; Lin, H.; Jiang, P.; Ding, J.; Guo, Z. Crystal structure and superoxide dismutase activity of a six-coordinate manganese (III) complex. Inorg. Chem. Commun. 2003, 6, 262–265. [Google Scholar] [CrossRef]
- Ribeiro, T.P.; Fernandes, C.; Melo, K.V.; Ferreira, S.S.; Lessa, J.A.; Franco, R.W.A.; Schenk, G.; Pereira, M.D.; Horn, A., Jr. Iron, copper and manganese complexes with in vitro superoxide dismutase and/or catalase activities that keep Saccharomyces cerevisiae cells alive under severe oxidative stress. Free Radical Biol. Med. 2015, 80, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Pap, J.S.; Kripli, B.; Bors, I.; Bogáth, D.; Giorgi, M.; Kaizer, J.; Speier, G. Transition metal complexes bearing flexible N3 or N3O donor ligands: Reactivity toward superoxide radical anion and hydrogen peroxide. J. Inorg. Biochem. 2012, 117, 60–70. [Google Scholar] [CrossRef]
- Durot, S.; Policar, C.; Cisnetti, F.; Lambert, F.; Renault, J.; Pelosi, G.; Blain, G.; Korri-Youssoufi, H.; Mahy, J. Series of Mn Complexes Based on N-Centered Ligands and Superoxide—Reactivity in an Anhydrous Medium and SOD-Like Activity in an Aqueous Medium Correlated to MnII/Mniii Redox Potentials. Eur. J. Inorg. Chem. 2005, 2005, 3513–3523. [Google Scholar] [CrossRef]
- Signorella, S.; Daier, V.; Ledesma, G.; Palopoli, C.; Back, D.F.; Lang, E.S.; Kopp, C.R.; Ebani, P.; Brum Pereira, M.; Giacomelli, C.; et al. Synthesis, structure and SOD activity of Mn complexes with symmetric Schiff base ligands derived from pyridoxal. Polyhedron 2015, 102, 176–184. [Google Scholar] [CrossRef]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Tocco, G.; Malfroy, E.; Adinolfi, C.A.; Kruk, H.; Baker, K.; Lazarowych, N.; Mascarenhas, J.; et al. Salen-manganese complexes as catalytic scavengers of hydrogen peroxide and cytoprotective agents: Structure-activity relationship studies. J. Med. Chem. 2002, 45, 4549–4558. [Google Scholar] [CrossRef]
- Ivanović-Burmazović, I.; van Eldik, R. Metal complex-assisted activation of small molecules. From NO to superoxide and peroxides. Dalton Trans. 2008, 5259–5275. [Google Scholar] [CrossRef]
- Liu, G.-F.; Dürr, K.; Puchta, R.; Heinemann, F.W.; van Eldik, R.; Ivanović-Burmazović, I. Chelate electronic properties control the redox behavior and superoxide reactivity of seven-coordinate manganese(II) complexes. Dalton Trans. 2009, 6292–6295. [Google Scholar] [CrossRef]
- Ramilo, C.A.; Laveque, V.; Guan, Y.; Lepock, J.R.; Tainer, J.A.; Nick, H.S.; Silverman, D.N. Interrupting the Hydrogen Bond Network at the Active Site of Human Manganese Superoxide Dismutase. J. Biol. Chem. 1999, 274, 27711–27740. [Google Scholar] [CrossRef]
- Borgstahl, G.E.O.; Parge, H.E.; Hickey, M.J.; Beyer, W.F.; Hallewell, R.A.; Tainer, J.A. The Structure of Human Mitochondrial Manganese Superoxide Dismutase Reveals a Novel Tetrameric Interface of Two 4-Helix Bundles. Cell 1992, 71, 107–118. [Google Scholar] [CrossRef]












| SBET (m2 g−1) | VµP (cm3 g−1) | VMP (cm3 g−1) | VTP (cm3 g−1) | wP (nm) | |
|---|---|---|---|---|---|
| SBA-15 | 568 | 0.06 | 0.51 | 0.63 | 5.2 |
| N3pn@OP-MS | 362 | 0.00 | 0.41 | 0.47 | 4.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richezzi, M.; Signorella, S.; Palopoli, C.; Pellegri, N.; Hureau, C.; Signorella, S.R. The Critical Role of Ligand Flexibility on the Activity of Free and Immobilized Mn Superoxide Dismutase Mimics. Inorganics 2023, 11, 359. https://doi.org/10.3390/inorganics11090359
Richezzi M, Signorella S, Palopoli C, Pellegri N, Hureau C, Signorella SR. The Critical Role of Ligand Flexibility on the Activity of Free and Immobilized Mn Superoxide Dismutase Mimics. Inorganics. 2023; 11(9):359. https://doi.org/10.3390/inorganics11090359
Chicago/Turabian StyleRichezzi, Micaela, Sharon Signorella, Claudia Palopoli, Nora Pellegri, Christelle Hureau, and Sandra R. Signorella. 2023. "The Critical Role of Ligand Flexibility on the Activity of Free and Immobilized Mn Superoxide Dismutase Mimics" Inorganics 11, no. 9: 359. https://doi.org/10.3390/inorganics11090359
APA StyleRichezzi, M., Signorella, S., Palopoli, C., Pellegri, N., Hureau, C., & Signorella, S. R. (2023). The Critical Role of Ligand Flexibility on the Activity of Free and Immobilized Mn Superoxide Dismutase Mimics. Inorganics, 11(9), 359. https://doi.org/10.3390/inorganics11090359