1. Introduction
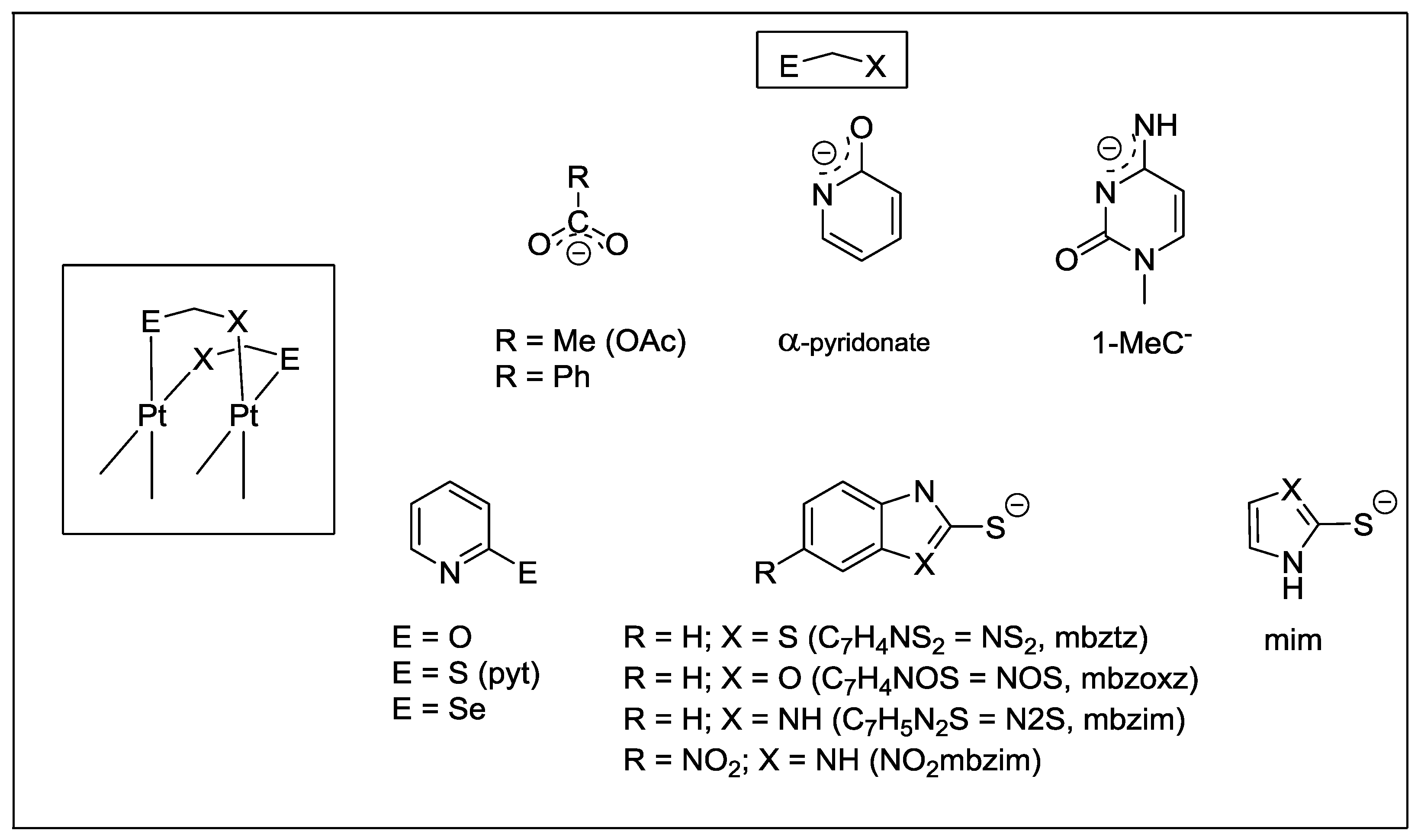
Half-lantern compounds of platinum with two four-bond bridging groups have been known for a long time (
Scheme 1). Cationic complexes derived from Cisplatin and analogues relevant to antitumor activity have been widely investigated over the last 40 years [
1]. Bridging ligands for dinuclear
cis-diaminoplatinum complexes include mainly carboxylate, α-pyridonate, amidates, and anionic nucleobases [
1,
2]. Some examples of divalent Pt
2(II,II) complexes are
cis-[Pt
2(μ-OAc)
2(NH
3)
4]
2+ [
3,
4],
ht-[Pt
2(μ-C
5H
4NO)
2(NH
3)
4]
2+ (C
5H
4NO = α-pyridonate) [
5] or
ht-
cis-[(NH
3)
2Pt(1-MeC
−-
N3,
N4)
2Pt(NH
3)
2](NO
3)
2 (1-MeC: 1-Methylcytosine) [
6], which exhibit Pt–Pt distances in the range 2.9–3.0 Å, indicating the absence of a formal metal-metal bond between the two d
8 metal ions [
7]. The dimers usually stack in the crystal lattice to form infinite chains [
1,
3,
4], with Pt–Pt separations of ~3.15 Å and hydrogen bonding between the oxygen atoms of the bridging ligands and the protons of the amine ligands, stabilizing the intermolecular interactions.
Scheme 1.
Schematic structures and four-bond bridging groups.
Scheme 1.
Schematic structures and four-bond bridging groups.
Neutral Pt
2 (II,II) complexes with bulkier monodentate ancillary ligands [
2] and carboxylate-bridging groups, such as [Pt
2(µ-OAc)
2(η
1-OAc)
2(PPh
3)
2] [
8], show a increase of the metal-metal distance of up to 3.10 Å. In spite of this, in [Pt
2Cl
2(µ-O
2CMe)
2(PMe
2Ph)
2] the
31P and
195Pt NMR data [
J(Pt–P') ≈ 30 Hz;
J(Pt–Pt') ≈ 1000 Hz)] strongly support
M···
M interactions [
9]. In complexes with pyridine-2-chalcogenolates (2-Py
E−,
E = O, S, Se) [
2], when
E = O, binuclear half-lantern complexes are formed [Pt
2Cl
2(µ-Py
E)
2(P
R3)
2] but no
M···
M interactions are detected from the NMR spectra. As the chalcogen size increases, monomeric complexes are predominantly formed. Four-bond ligands with a larger bite size, such as xanthates, dithiocarbamates, or dithiophosphates, render exclusively mononuclear complexes [
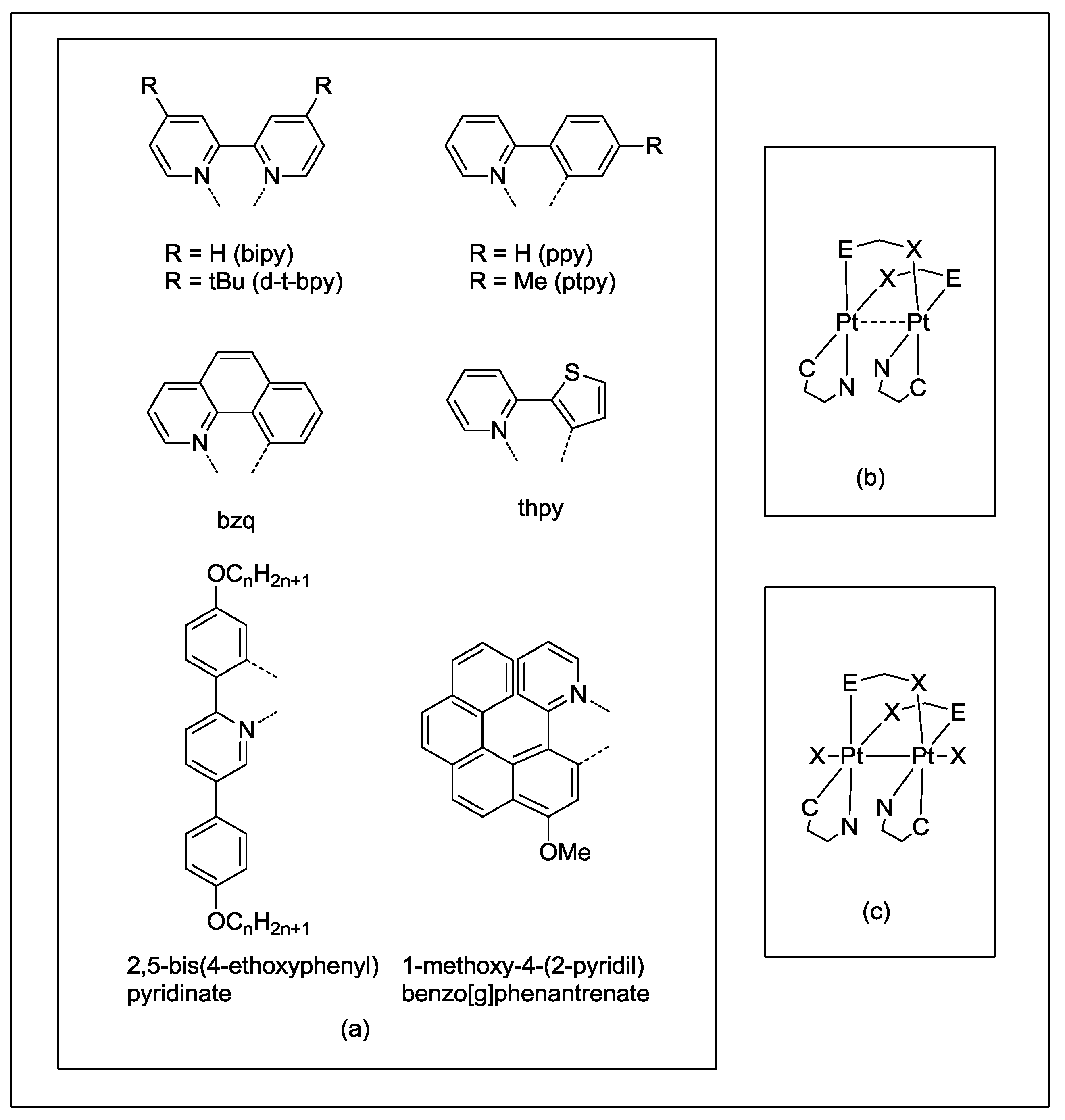
2]. Therefore, the metal-metal separation is significantly affected by the bite size and orientation of the bridging ligands, as well as by the bulkiness of the ancillary ones. Planar di- or tert-dentated polyimines and cyclometalated ligands do not hinder the face-to-face orientation of the metal planes, allowing metal-metal interactions to exist (
Scheme 2a,b). Many of these half-lantern compounds exhibit luminescence from triplet metal-metal-to-ligand charge transfer (
3MMLCT) excited states. The MMLCT transition involves charge transfer between a filled Pt–Pt antibonding (dσ*) orbital and an empty ligand-based π* orbital [dσ*(Pt)
2→π*(
L)] of the polyimine or the cyclometalated ligand.
Scheme 2.
Schematic Pt2(II,II) and Pt2(III,III) structures with diimines and cyclometalated ligands.
Scheme 2.
Schematic Pt2(II,II) and Pt2(III,III) structures with diimines and cyclometalated ligands.
Considering half-lantern complexes containing diimines, Kato and co-workers were able to isolate and characterize the
syn and
anti isomers of [Pt
2(pyt)
2(bipy)
2][PF
6]
2 from the mixture of both, obtained from [PtCl
2(bipy)], pyridine-2-thiol and NH
4PF
6 [
10]. The
syn isomer has a head-to-headconfiguration of two bridging pyt and Pt···Pt separation of 2.923(1) Å. The
anti isomer shows a head-to-tail arrangement of them and inter-metallic distance of 2.997(1) Å.
Tzeng and co-workers prepared the dinuclear diimine complexes
syn- or
anti-[Pt
2(μ-NS)
2(dtbpy)
2]
2+ dtbpy = 4,4'-di-tert-butyl-2,2'-bipyridine, NHS = pyridine-2-thiol (Hpyt), 2-mercaptobenzothiazole (HNS
2), 2-mercaptobenzimidazole (HN
2S), 2-mercaptobenzoxazole (HNOS)) by reaction of the mononuclear derivative [Pt(dtbpy)Cl
2] with the corresponding HNS in the presence of NaOMe [
11,
12]. The
anti-[Pt
2(pyt)
2(dtbpy)
2]
2+ isomer shows a Pt···Pt distance of 2.917(2) Å and also shows photoluminescence at room temperature in the solid state (
λmax = 606 nm) [
12]. The
syn- or
anti-[Pt
2(μ-NS)
2(dtbpy)
2]
2+ (NS = NS
2, N
2S, NOS) exhibit intramolecular Pt···Pt distances of 2.9727–3.0079 Å, which lead to Pt···Pt and π-π interactions in the solid state and, additionally, intermolecular Pt···Pt and π-π contacts in the
syn isomers.
Changing diimines by
C,
N-cyclometalated fragments allowed neutral half-lantern Pt(II) complexes, such as complexes [Pt
2(C^N)
2(pyt)
2] (Hpyt = pyridine-2-thiol, HC^N = 2-phenylpyridine (Hppy), 2-(
p-tolyl)pyridine (Hptpy), 2-(2-thienyl)pyridine (Hthpy), benzo[h]quinolone (Hbzq)) to be prepared from (NBu
4)[Pt(C^N)Cl
2] and Hpyt with an excess of tributylamine (NBu
3) [
13,
14]. These dinuclear complexes have a rigid dinuclear framework and only exhibit head-to-tail conformation due to the different
trans influence of C and N of the C^N ligand, whereas the pyt group prefers the
N-coordination at the
trans position to C (
Scheme 2b). Complexes [Pt
2(C^N)
2(pyt)
2] (C^N = ppy, ptpy, thpy, bzq) display short Pt···Pt distance (2.82–2.88 Å), suggesting the existence of strong Pt···Pt interactions, and exhibit red luminescence at room temperature both in solid state (λ
max = 633–711 nm) and in solution (λ
max = 650–715 nm) derived from a
3MMLCT origin.
The half-lantern Pt(II) complexes [{Pt(bzq)(μ-C
7H
4NS
2-κN,S)}
2]·Me
2CO and [{Pt(bzq)(μ-C
7H
4NOS-κN,S)}
2] (Hbzq: benzo[h]quinolone; C
7H
4NS
2: 2-mercaptobenzothiazolate, C
7H
4NOS: 2-mercaptobenzoxazolate) were prepared selectively (yield
ca. 80%) by reaction of [Pt(bzq)(NCMe)
2]ClO
4 and KC
7H
4N
YS (
Y = S, O) in 1:1 molar ratio in acetone/methanol (2:1) at room temperature [
15,
16]. Complex [{Pt(bzq)(μ-C
7H
4NOS-κN,S)}
2] and the analogous [{Pt(ppy)(μ-C
7H
4NOS-κN,S)}
2] were also obtained by one-pot reaction between [{Pt(C^N)(µ-Cl)}
2] (C^N, bzq, ppy) and NOSH in a ratio of 1.0:4.2 in THF at r.t. in the presence of NaOAc in yields of 32%–35% [
17]. All three complexes show an
anti configuration, with the N of the bridging groups coordinated at the
trans position to C, and intermetallic distances of 2.91 Å, 2.97 Å (bzq) and 3.02 Å (ppy). The bzq complexes show significant π-π interactions that are absent in the ppy one. These compounds were isolated as orangish-red solids, which exhibit a weak absorption at ~500 nm and intense orangish-red photoluminescence at room temperature both in solid state (λ
max = 665–691 nm) and in toluene solution (λ
max = 660–677 nm) [
15,
16]. TD-DFT studies on complexes [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)}
2] (
Y = S, O) [
15,
16] proved the
1,3MMLCT character of their lower energy absorption and emission, which is affected by the strength of the π···π interactions. Complexes [{Pt(pbt)(μ-C
7H
4NYS-κN,S)}
2] (pbt: 2-phenylbenzothiazole;
Y = S: 2-mercaptobenzothiazolate,
Y = O: 2-mercaptobenzoxazolate) were prepared later by a similar procedure with yields of 60%–70% [
18].
The half-lantern Pt
2(II,II) complexes show complex and interesting redox chemistry [
19]. Oxidation of
cis-diaminoplatinum Pt
2 (II,II) leads to oligomeric mixed-valence “platinum blues” with oxidation states varying +2.25, +2.5 and 3. Apart from these, discrete
cis-diaminoplatinum (III) dimers, such as,
ht-[Pt
2(μ-C
5H
4NO)
2(NH
3)
4X2]
2+ (C
5H
4NO = α-pyridonate,
X = NO
3, NO
2, Cl, Br) [
20,
21],
ht-
cis-[(
OH
2)(NH
3)
2Pt(1-MeC
−-
N3,
N4)
2-Pt(NH
3)
2(
OH
2)] (ClO
4)
4·H
2O [
22], or
cis-[Pt
2(μ-OAc)
2X2(NH
3)
4]
2+ (
X = Cl, Br) [
4] are known, with bond distances of ~2.6 Å. The diamagnetic nature of these compounds and the shortening of the Pt−Pt distance with respect to the Pt
2(II,II) are consequences of the formation of a metal-metal σ bond.
Oxidation of C,
N-cyclometalated Pt
2(II,II) compounds do not lead to mixed-valence oligomeric species but rather to discrete Pt
2(III,III) dimers [
13] (
Scheme 2c). By way of example, complexes [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)}
2] (
Y = S, O) undergo two-center, two-electron oxidation in their reactions with halogens (
X2 = Cl, Br, I) [
15,
16] to give [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)
X}
2] (
Y = S, O;
X = Cl, Br, I) which show Pt−Pt distances (2.63 to 2.68 Å), about 10% shorter than their starting complexes (2.91, 2.97 Å). The half-lantern Pt
2(III,III)X
2 compounds (
X = Cl, Br, I) seem to be quite stable, being the final products of diverse kinds of reactions. The dichloro compounds [Pt
2(C^N)
2(µ-pyt)
2Cl
2] (Hpyt = pyridine-2-thiol, HC^N = 2-phenylpyridine (Hppy), 2-(
p-tolyl)pyridine (Hptpy), 2-(2-thienyl)pyridine (Hthpy), benzo[h]quinolone (Hbzq)) were the final products of the reactions between (NBu
4)[Pt(C^N)Cl
2] and Hpyt in the absence of a base [
13]; otherwise, in the presence of NBu
3, the divalent compounds [Pt
2(C^N)
2(µ-pyt)
2] were formed. Compound [Pt
2(ppy)
2(µ-pyt)
2Cl
2], which shows a Pt-Pt distance of 2.6150(8) Å, was also obtained by reaction of the corresponding Pt
2(II,II) derivative with HCl or by recrystallization of Pt
2(II,II) from chloroform [
14].
Pt
2(III,III)Cl
2 derivatives can also be obtained by using the dinuclear complexes [{Pt(C^N)(µ-Cl)}
2] as starting materials. Examples of this synthetic route are the synthesis of [{Pt(ppy)(µ-N^S)Cl}
2] (HN^S = 2-mercaptobenzimidazole (mbzimH), 5-nitro-2-mercaptobenzimidazole (NO
2-mbzimH), 2-mercaptobenzothiazole (mbztzH), 2-mercaptobenzoxazole (mbzoxzH), 2-mercaptoimidazoline (mimH)) by reaction of [{Pt(C^N)(µ-Cl)}
2] and HN^S in CHCl
3 at room temperature [
23]. The Pt−Pt distances in these trivalent dinuclear complexes range from 2.6185(14) Å to 2.6445(3) Å.
Carboxylate-bridged Pt
2(III,III)Cl
2 derivatives such as [{Pt(C^N)(µ-O
2C
R)Cl}
2] (C^N = 2,5-bis(4-ethoxyphenyl)pyridine-H,
R = Me [
24], 1-methoxy-4-(2-pyridyl)benzo[
g] phenantrene-H,
R = Ph [
25,
26] could be obtained by reaction of the mononuclear [Pt(C^N)Cl(DMSO)] with acetic acid [
24] or by reaction of the dinuclear [{Pt(C^N)(µ-Cl)}
2] with AgO
2CPh [
25,
26]. The metal-metal distances in these di-μ-carboxylate complexes are 2.5730(3) Å [
24] and 2.5952(6) Å [
26], both of which are quite shorter than those observed in the above described complexes containing two N^S bridging groups.
None of the described half-lantern Pt
2(III,III) complexes are luminescent in the visible region. In general, the d
7−d
7 complexes are no emitter, both in solution and in solid state, there being just a few exceptions, for example, [Pt
2(µ-pop)
4X
2]
4− (pop =
P,
P-pyrophosphite,P
2O
5H
22−,
X = Cl, Br, SCN, or py) [
27,
28], which exhibit red luminescence in an alcohol glass or in solid state at low temperature and [{Pt(κ
2-As,C-C
6H
3-5-CHMe
2-2-AsPh
2)
2X}
2] (
X = Cl, Br, I, CN) [
29] that emit in the visible to NIR region even at room temperature.
In the course of our research on half-lantern Pt(II) complexes, we have prepared a new Pt
2(II,II) derivative, [{Pt(bzq)(μ-L)}
2] [Hbzq = benzo[h]quinolone, HL = CF
3C
4H
2N
2SH: 4-(trifluoromethyl)pyrimidine-2-thiol] and the two-electron-oxidized dihalodiplatinum (III) complexes [{Pt(bzq)(μ-
L)
X}
2] (
L = CF
3C
4H
2N
2S-κN,S;
X: Cl, Br, I). In spite of the similarities of [{Pt(bzq)(μ-CF
3C
4H
2N
2S-κN,S)}
2] with compounds [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)}
2] (
Y = S, O) [
15,
16], it showed not luminescence in the visible region and, as expected, the Pt
2(III,III)
X2 neither did.
2. Results and Discussion
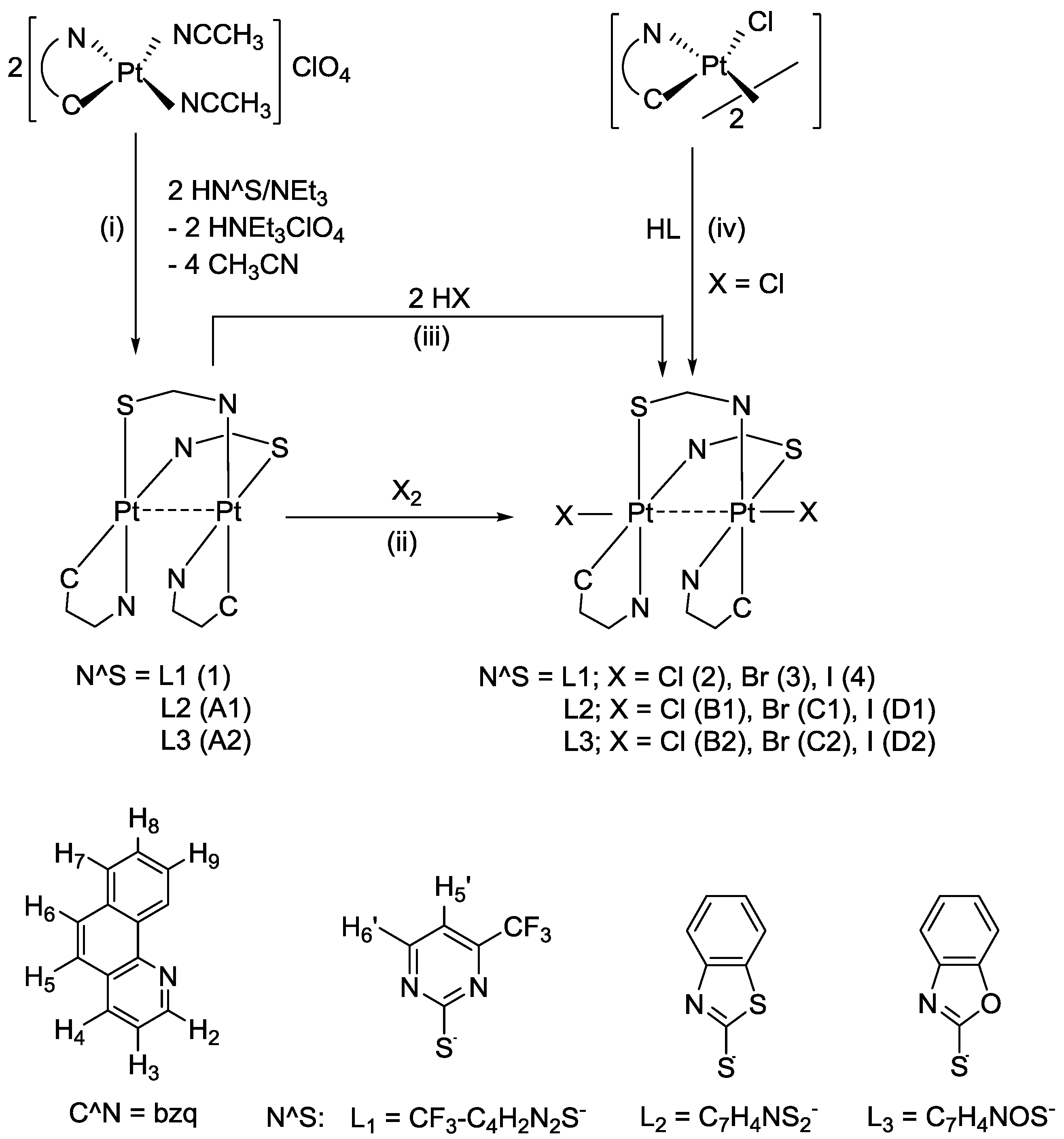
The divalent complex [{Pt(bzq)(μ-
L)}
2] (
1) [Hbzq = benzo[h]quinolone, H
L = CF
3C
4H
2N
2SH: 4-(trifluoromethyl)pyrimidine-2-thiol] was obtained by refluxing equimolar amounts of [Pt(bzq)(NCMe)
2]ClO
4 and 4-(trifluoromethyl)pyrimidine-2-thiol with an excess of NEt
3 in acetone-methanol. Compound
1 precipitated in the reaction mixture from which it was separated and obtained as a pinkish-red, air-stable solid in a good yield (92%) (see
Scheme 3, path i, and
Experimental section). No single crystals of
1 could be grown for X-ray purposes, but evidences from other techniques lead us to propose the structure represented in
Scheme 3. The dinuclear nature of
1 is revealed from its mass spectrum that shows two important peaks at
m/
z 1105.1 (97%) and 925.2 (100%) corresponding to [M]
+ and ([M-CF
3-C
4H
2N
2S]
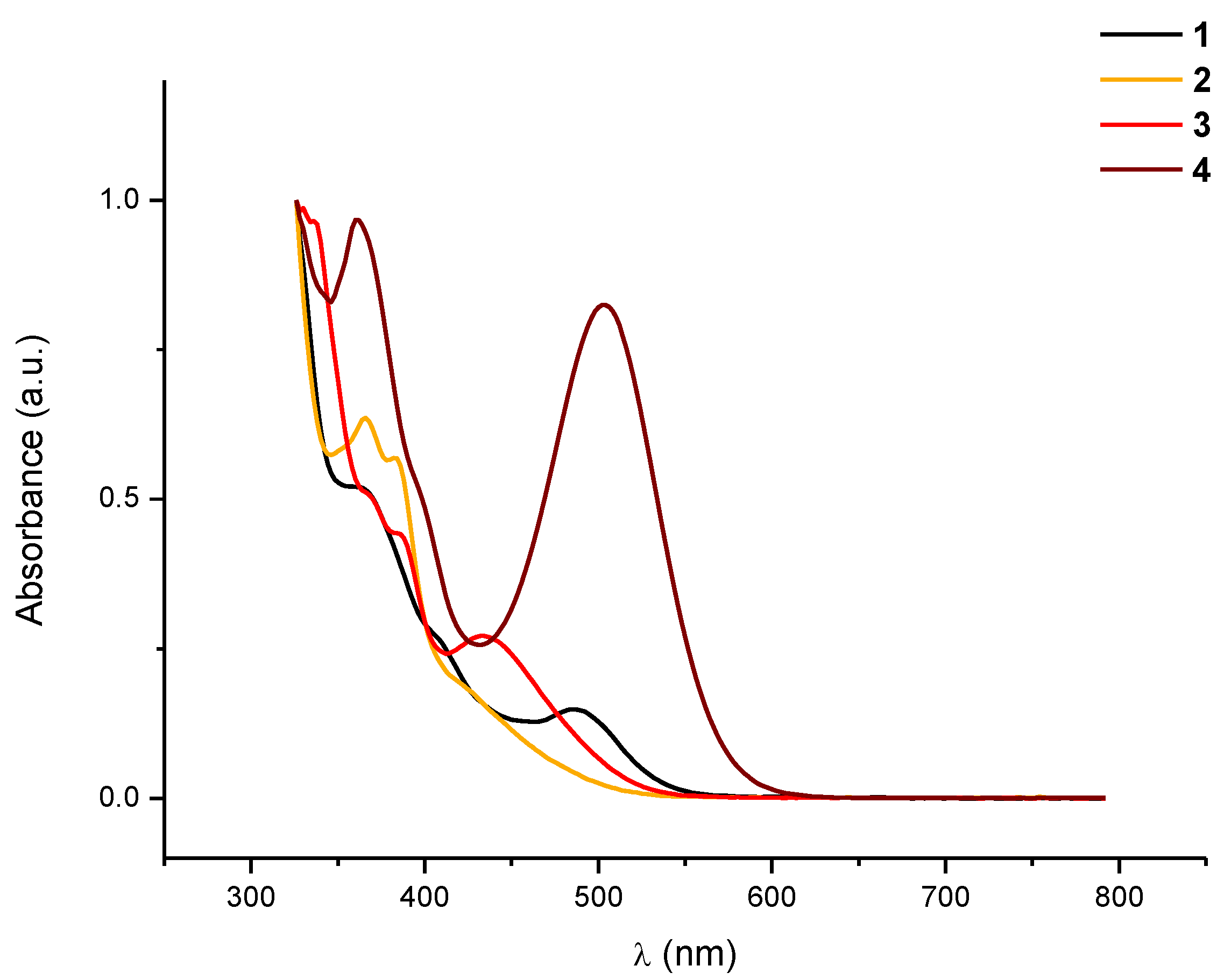
+ respectively. The electronic absorption spectrum of
1 recorded in CH
2Cl
2 (
Figure 1) also provided some structural information. It shows a low intensity band centered at 486 nm (CH
2Cl
2), which can be tentatively assigned to a metal-metal-to-ligand charge transfer transition, (
1MMLCT) [dσ*(Pt)
2→π*(bzq)] as in the analogous half-lantern compounds [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)}
2] (
Y = O, 480 nm CH
2Cl
2;
Y = S, 487 nm CH
2Cl
2) [
15,
16]. The presence of this band due to Pt···Pt interactions is indicative of the existence of two platinum centers located in close proximity. The rigidity of the half-lantern structure would allow the preservation of these interactions in solution. The presence of only one set of signals corresponding to the bzq and the bridging CF
3C
4H
2N
2S-κN,S ligands, indicates that compound
1 exists as one single, symmetric isomer, most probably the
anti one, as observed in the previously reported analogous compounds [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)}
2] (
Y = O, S). The scarce solubility of
1 in most common solvents precludes obtaining more information about its structure in solution by other means, such as a NOE spectrum.
Scheme 3.
Reaction scheme and numbering for NMR purposes.
Scheme 3.
Reaction scheme and numbering for NMR purposes.
As expected from the presumed short Pt−Pt distance, compound
1 undergoes two-electron oxidation upon treatment with halogens
X2 (
X2 = Cl
2, Br
2 or I
2) to give the corresponding dihalodiplatinum (III) complexes [{Pt(bzq)(μ-
L)
X}
2] (
L = CF
3C
4H
2N
2S-κN,S;
X: Cl
2, Br
3, I
4) as yellowish-orange, orange and pinkish-red solids, respectively, in very high yield (
Scheme 3, path ii and
Experimental section). The IR and
1H-NMR spectra of
2–
4 are very similar and show small changes with respect to the starting complex
1 ones; the different colors of these complexes are revealed in their UV-vis spectra (
Figure 1). The X-ray studies confirmed that these Pt(III) complexes are isostructural (see below). Complexes
2–
4 were also obtained by reaction of
1 with H
X (molar ratio 1:2, 10% excess of H
X) in THF with yields of about 80% (
Scheme 3 path iii and
Experimental section).
Figure 1.
Normalized absorption spectra in CH2Cl2 solution (10−4 M) of 1–4 at room temperature.
Figure 1.
Normalized absorption spectra in CH2Cl2 solution (10−4 M) of 1–4 at room temperature.
Compound
2 was also obtained by reaction of [{Pt(bzq)(μ-Cl)}
2] with HL (4-(trifluoromethyl)pyrimidine-2-thiol) in molar ratio 1:2 in THF (
Scheme 3 path iv and
Experimental section). This pathway rendered compound
2 with a moderate yield, as previously observed in the synthesis of the analogous compounds [{Pt(2-PhPy)(μ-
L)Cl}
2] (H
L = 2-mercaptobenzimidazole, 5-nitro-2-mercaptobenzimidazole, 2-mercaptobenzothiazole, 2-mercaptobenzoxazole, 2-mercaptoimidazoline) [
23]. The use of [{Pt(bzq)(μ-Cl)}
2] as starting material, has important limitations: (a) it exclusively renders the dichloro compound, preventing comparison of dichloro with dibromo and diiodo derivatives and (b) it proceeds with low yield, probably because side reactions occurred simultaneously.
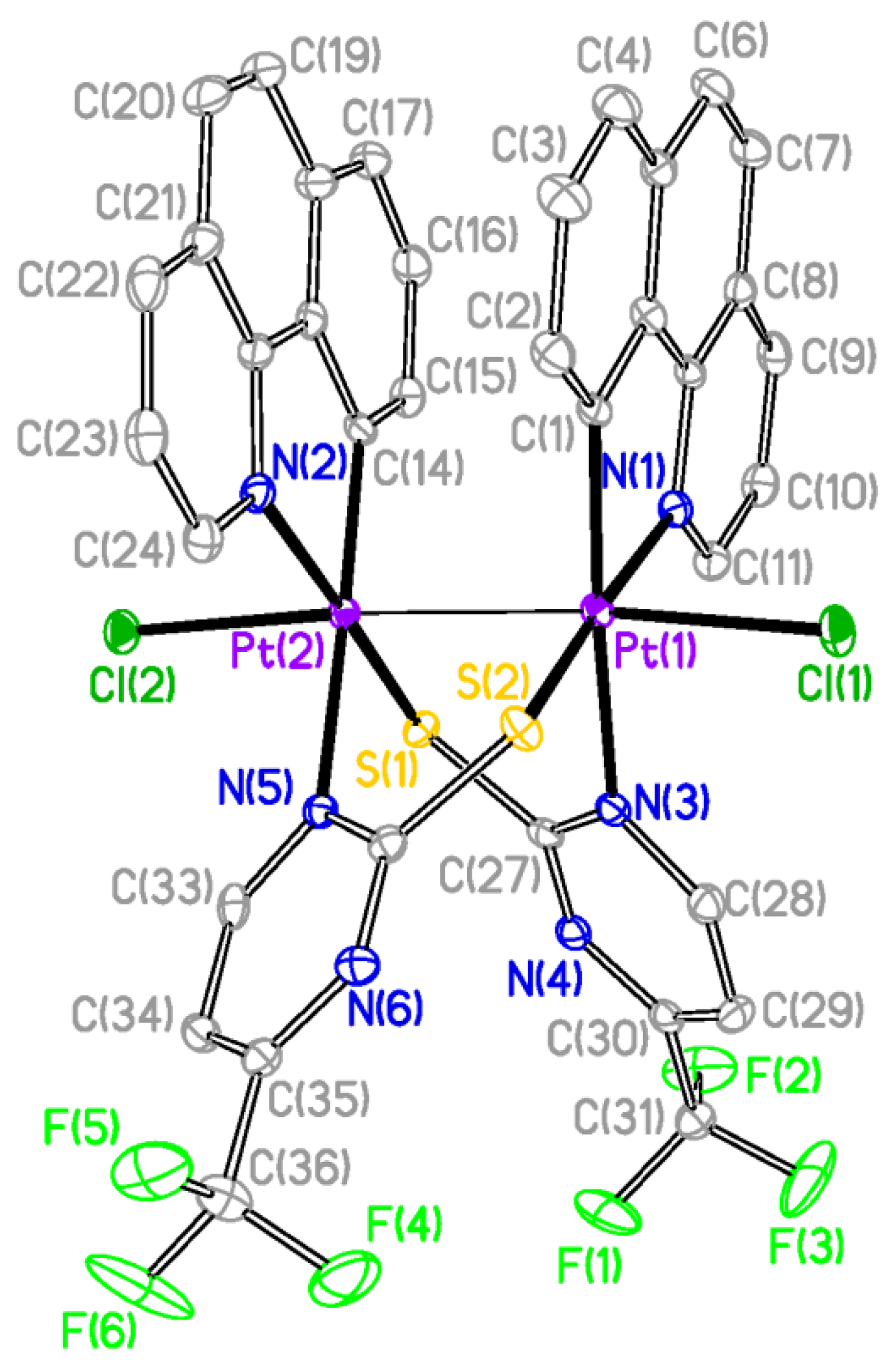
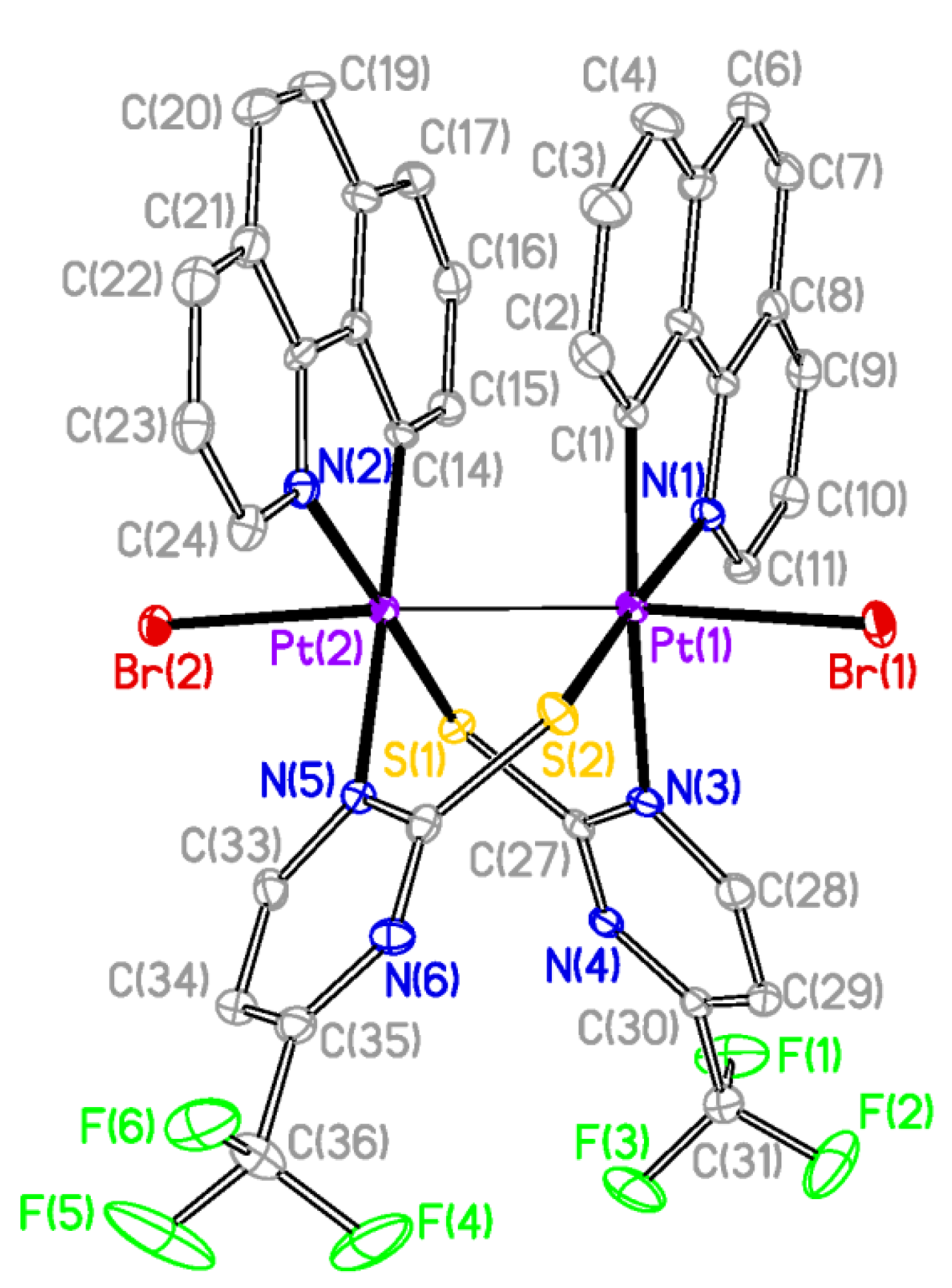
The X-ray structures of
2 and
3 are shown in
Figure 2 and
Figure 3 respectively and a selection of bond distances and angles is listed in
Table 1. They confirmed the expected half-lantern structure and the
anti configuration of the molecule. Each Pt (III) center has a distorted octahedral environment with the axial positions occupied by an halogen atom (Cl
2, Br
3) and the other Pt(III) center, and with the X−Pt−Pt angles being close to 174°. Compounds
2 and
3 show Pt-Pt distances (2.61188(15) Å
2, 2.61767(16) Å
3), similar to that of [Pt
2(ppy)
2(µ-pyt)
2Cl
2] (2.6150(8) Å) [
14], all three being in the low range of those observed in Pt
2(III,III)X
2 half-lantern complexes [
15,
16]. The shortening of the Pt-Pt distance in
2 and
3 with respect to those of the analogous compounds (
X = Cl: 2.6420(3) Å
B1, 2.6383(3) Å
B2; Br: 2.6435(4) Å
C1, 2.6671(9) Å
C2) [
15,
16] seems to be related to the NCS angle values of the 4-membered bridges. These angles are close to 120° in
2 and
3 (120.47(22)°
2, 121.23(24)°
3) and clearly smaller than those in
B1(128.4(4)°),
C1(129.03(52)°),
B2 (128.2(5)°) and
C2 (131.56(00)°). In addition, the Pt-Pt distances in
2 and
3 were shorter in the dichloro than in the dibromo derivative, in accordance with the larger
trans-influence of Br with respect to Cl [
30], as was previously observed in [Pt
2(P
2O
5H
2)
4X2]
4− (
X = Cl, Br, I) [
31], [Pt
2(µ-κAs,κC-C
6H
3-5-CHMe
2-2-AsPh
2)
4X2] (
X = Cl, Br, I) [
29], [Pt
2(µ-κAs,κC-C
6H
3-5-Me-2-AsPh
2)
4X2] (
X = Cl, Br, I) [
32] or [{Pt(bzq)(μ-C
7H
4N
YS-κN,S)
X}
2] (
Y = O, S;
X = Cl, Br, I) [
15,
16]. The Pt(III)−
X distances are in the range of those found in other complexes with these kinds of ligands. In these dinuclear Pt
2(III,III)
X2 complexes, the two platinum coordination planes are almost parallel, with a small interplanar angle [9.79(5)° 2, 9.76(6)° 3] and the Pt-Pt line is almost perpendicular to the two Pt square coordination planes, the biggest angle being 5.44(4)° (Pt1−Pt2 line and Pt1 plane for compound
2). The crystal packing of these two complexes does not show intermolecular interactions among neighbor molecules.
Table 1.
Selected Bond Distances (Å) and angles (deg) of 2 and 3.
Table 1.
Selected Bond Distances (Å) and angles (deg) of 2 and 3.
| Bonds and angles | 2 CH2Cl2 | 3·CH2Cl2 |
|---|
| Pt(1)−C(1) | 2.015(3) | 2.013(3) |
| Pt(1)−N(1) | 2.090(2) | 2.092(2) |
| Pt(1)−Nμ-N^S | 2.151(2) | 2.165(3) |
| Pt(1)−Sμ-N^S | 2.3016(7) | 2.2995(8) |
| Pt(1)−
X(1) | 2.4305(7) | 2.5593(3) |
| Pt(1)−Pt(2) | 2.61188(15) | 2.61767(16) |
| Pt(2)−C(14) | 2.015(3) | 2.015(3) |
| Pt(2)−N(2) | 2.078(2) | 2.080(3) |
| Pt(2)−Nμ-N^S | 2.151(2) | 2.156(3) |
| Pt(2)−Sμ-N^S | 2.3036(7) | 2.3047(8) |
| Pt(2)−
X(2) | 2.4381(7) | 2.5692(3) |
| C(1)−Pt(1)−N(1) | 81.44(11) | 81.55(11) |
| N(1)−Pt(1)−Nμ-N^S | 93.43(9) | 93.37(10) |
| C(1)−Pt(1)−Sμ-N^S | 96.52(8) | 96.25(9) |
| Nμ-N^S−Pt(1)−Sμ-N^S | 88.46(6) | 88.70(7) |
| C(14)−Pt(2)−N(2) | 81.68(11) | 81.56(12) |
| N(2)−Pt(2)−Nμ-N^S | 93.87(9) | 93.69(10) |
| C(14)−Pt(2)−Sμ-N^S | 96.35(9) | 96.31(9) |
| Nμ-N^S−Pt(2)−Sμ-N^S | 88.13(7) | 88.48(7) |
| C(1)−Pt(1)−
X(1) | 86.94(8) | 86.29(9) |
| N(1)−Pt(1)−
X(1) | 88.94(6) | 88.73(7) |
| Nμ-N^S−Pt(1)−
X(1) | 91.44(6) | 92.14(7) |
| Sμ-N^S−Pt(1)−
X(1) | 86.47(2) | 86.71(2) |
| Pt(2)−Pt(1)−
X(1) | 173.389(18) | 173.901() |
| C(14)−Pt(2)−
X(2) | 89.04(8) | 88.22(9) |
| N(2)−Pt(2)−
X(2) | 88.51(6) | 88.71(7) |
| Nμ-N^S−Pt(2)−
X(2) | 91.36(6) | 92.34(7) |
| Sμ-N^S−Pt(2)−
X(2) | 88.25(2) | 88.09(2) |
| Pt(1)−Pt(2)−
X(2) | 175.352(1) | 175.567(9) |
Differently to the analogous compounds [{Pt(bzq)(μ-C7H4NOS-κN,S)}2] (A1) and [{Pt(bzq)(μ-C7H4NS2-κN,S)}2] (A2), compound 1 showed no luminescence either in solid or in solution and, as expected, neither did the d7–d7 compounds 2–4.
Figure 2.
Molecular Structure of compound 2. Ellipsoids are drawn at their 50% probability level; solvent molecules and hydrogen atoms were omitted for clarity.
Figure 2.
Molecular Structure of compound 2. Ellipsoids are drawn at their 50% probability level; solvent molecules and hydrogen atoms were omitted for clarity.
Figure 3.
Molecular Structure of compound 3. Ellipsoids are drawn at their 50% probability level; solvent molecules and hydrogen atoms were omitted for clarity.
Figure 3.
Molecular Structure of compound 3. Ellipsoids are drawn at their 50% probability level; solvent molecules and hydrogen atoms were omitted for clarity.
3. Experimental section
General procedures and materials. Elemental analyses were carried out with a Perkin Elmer (Waltham, MA, USA) 2400 CHNS analyzer. IR spectra were recorded on a Perkin-Elmer (Waltham, MA, USA) Spectrum 100 FT-IR Spectrometer (ATR in the range 250–4000 cm−1). Mass spectral analyses were performed with a Microflex MALDI-TOF Bruker or an Autoflex III MALDI-TOF Bruker (Madison, WI, USA), instruments. NMR spectra were recorded on a Bruker (Madison, WI, USA) AV-400 spectrometer using the standard references: SiMe4; J is given in Hz and assignments are based on 1H-1H-COSY experiments. Absorption spectra were recorded on a Thermo Electron Corporation (Waltham, MA, USA) evolution 600 spectrophotometer and diffuse reflectance UV-vis (DRUV) spectra were recorded on a Thermo Electron Corporation (Waltham, MA, USA) Evolution 600 spectrophotometer equipped with a Praying Mantis integrating sphere. The solid samples were homogeneously diluted with silica. The mixtures were placed in a homemade cell equipped with quartz window.
The starting material [Pt(bzq)(NCMe)
2]ClO
4 was prepared, as described elsewhere [
33]. (CF
3)C
4H
2N
2SH was used as purchased from Aldrich (St. Louis, MO, USA).
[{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)}2] (1). A yellowish-orange suspension of [Pt(bzq)(NCMe)2]ClO4 (0.251 g, 0.453 mmol) in acetone (20 mL) was treated with a solution of (CF3)C4H2N2SH (0.082 g, 0.453 mmol) in methanol (10 mL) and NEt3 (0.5 mL). The mixture was stirred and refluxed for 2 h and then was concentrated to about 15 mL. The resulting pinkish-red solid was filtered-off, washed with MeOH (2 × 3 mL) and Et2O (2 × 5 mL) and dried, 1. Yield: 0.231 g, 92%. Anal. Calcd for C36H20F6N6Pt2S2: C, 39.13; H, 1.82; N, 7.61; Found: C, 39.10; H, 1.99; N, 7.55; 1H-NMR (CD2Cl2, 400.16 MHz, 298K): δ 9.10 (2H, d, 3J(H6',H5') = 5.7; H6’), 7.97 (2H, dd, 3J(H4,H3) = 7.9, 4J(H4,H2) = 1.0, H4), 7.93 (2H, dd, 3J(H2,H3) = 5.2; H2), 7.32 (2H, dd, H3), 7.20 (4H, m, H9, H5'), 7.16 (4H, AB, 3J(H5,H6) = 8.6, H5, H6), 6.86 (2H, d, 3J(H7,H8) = 7.5; H7), 6.64 (2H, dd, 3J(H8,H9) = 7.5, H8) ppm; 19F-NMR (CD2Cl2, 376 MHz, 298K): δ −70.9 (s, CF3) ppm; MS (MALDI+): m/z 925.2 ([M − CF3-C4H2N2S]+, 100%), 1105.1 (M+, 97%); IR (cm−1): 1620 d, 1575 m, 1545 m, 1450 d, 1426 m, 1405 m, 1328 f, 1314 mf, 1205 f, 1178 m, 1144 f, 1113 mf, 1087 f, 1053 m, 998 m, 928 m, 841 f, 823 f, 762 m, 751 m, 730 f, 710 f, 671 f, 654 m, 526 m, 469 m, 288 m, 281 m, 264 m.
[{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)Cl}2] (2). (Method A) A solution of Cl2 in CCl4 0.25M (0.602 mL, 0.151 mmol) was added to a pinkish-red suspension of 1 (0.1513 g, 0.137 mmol) in THF (4 mL). The resulting orange mixture was stirred for 4 h and concentrated to 2 mL. Addition of Et2O (5 mL) to the residue rendered a yellowish-orange solid that was filtered-off and washed with Et2O (2 × 4 mL), 2. Yield: 0.134 g, 83%. Anal. Calcd for C36H20Cl2F6N6Pt2S2: C, 36.77; H, 1.71; N, 7.15; Found: C, 36.65; H, 2.06; N, 7.08; 1H-NMR (CD2Cl2, 400.16 MHz, 298K): δ 10.00 (2H, d, 3J(H6',H5') = 6.1; H6'), 8.13 (2H, dd, 3J(H2,H3) = 5.6, 4J(H2,H4) = 1.0, 3J(Pt,H2) = 22.5, H2), 8.06 (2H, dd, 3J(H4,H3) = 8.1, 4J(H4,H2) = 1.0, H4), 7.55 (2H, dd, H3), 7.40 (2H, d, H5’), 7.29 (4H, AB, 3J(H5,H6) = 8.8, H5, H6), 7.04 (2H, dd, 3J(H9,H8) = 7.5; 4J(H9,H7) = 0.8 Hz, 3J(Pt,H9) = 31.6, H9), 6.93 (2H, d, 3J(H7,H8) = 8.0; H7), 6.75 (2H, dd, H8) ppm; 19F-NMR (CD2Cl2, 376 MHz, 298K):δ −70.6 (s, CF3) ppm. IR (cm−1): 1621 d, 1586 m, 1575 m, 1549 m, 1454 d, 1436 m, 1407 m, 1343 f, 1332 f, 1322 mf, 1205 f, 1185 m, 1142 f, 1116 mf, 928 m, 844 m, 830 mf, 819 m, 764 m, 731 f, 711 f, 674 f, 654 m, 514 m, 467 m, 438 m. (Method B) A solution of HCl in H2O 1M (299 µL, 0.299 mmol) was added to a pinkish-red suspension of 1 (0.150 g, 0.136 mmol) in THF (10 mL) and the mixture was stirred for 51 h in the darkness. During this time, a yellowish-orange solid was precipitated. The solvent was evaporated to ca. 2 mL and Et2O (10 mL) was added to the reaction mixture. The yellowish-orange solid 2·THF was filtered-off, washed with Et2O (2 × 5 mL) and dried to the air. Yield: 0.1368 g, 81%. (Method C) A yellow solution of C5H3N2F3S (0.062 g, 0.348 mmol) in THF (20 mL) was added drop by drop to a yellow suspension of [{Pt(bzq)(µ-Cl)}2] (0.142 g, 0.174 mmol) in THF (10 mL). Then, the mixture was stirred for 70 h to give a yellow solution that was evaporated to dryness. Addition of acetone (15 mL) to the residue rendered a yellowish- orange solid, 2, that was filtered-off, washed with acetone (2 × 5 mL) and Et2O (2 × 5 mL) and dried to the air. Yield: 0.0341 g, 24%.
[{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)Br}2] (3). (Method A) was prepared in the same way as 2, but using Br2 (0.149 mmol 7.66 µL, d = 3.119 g/mL), 1 (0.150 g, 0.136 mmol). Orange solid 3, Yield: 0.154 g, 90%. Anal. Calcd for Br2C36H20F6N6Pt2S2: C, 34.19; H, 1.59; N, 6.65; Found: C, 33.80; H, 1.61; N, 6.71. 1H-NMR (CD2Cl2, 400.16 MHz, 298K): δ 10.14 (2H, d, 3J(H6',H5') = 6.0; H6'), 8.19 (2H, dd, 3J(H2,H3) = 5.5, 4J(H2,H4) = 1.3, 3J(Pt,H2) = 22.2, H2), 8.05 (2H, dd, 3J(H4,H3) = 7.8,4J(H4,H2) = 1.3, H4), 7.56 (2H, dd, H3), 7.38 (2H, d, H5'), 7.29 (4H, AB, 3J(H5,H6) = 8.8 Hz, H5, H6), 7.00 (2H, dd, 3J(H9,H8) = 7.6; 4J(H9,H7) = 0.7, 3J(Pt,H9) = 31.7, H9), 6.90 (2H, d, 3J(H7,H8) = 8.0; H7), 6.74 ppm (2H, dd, H8); 19F-NMR (CD2Cl2, 376 MHz, 298K):δ −70.6 (s, CF3) ppm. IR (cm−1): 1621 d, 1586 m, 1575 m, 1549 m, 1454 m, 1436 m, 1407 m, 1343 f, 1332 f, 1322 mf, 1205 f, 1185 f, 1142 f, 1116 mf, 1053 m, 928 m, 844 f, 830 mf, 819 f, 764 f, 731 f, 711 f, 674 f, 654 m, 514 m, 467 m, 438 m, 271 m. (Method B) A solution of HBr in H2O 1M (296 µL, 0.296 mmol) was added to a pinkish-red suspension of 1 (0.149 g, 0.135 mmol) in THF (20 mL) and the mixture was stirred for 72 h in the darkness, while an orange solid was precipitated. The solvent was evaporated to ca. 2 mL and Et2O (10 mL) was added to the reaction mixture. The orange solid 3·THF was filtered-off, washed with Et2O (2 × 5 mL) and dried to the air. Yield: 0.127 g, 70%.
[{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)I}2] (4). (Method A) was prepared in the same way as 2, but using a solution of I2 (0.038 g, 0.151 mmol) in THF (4 mL), a suspension of 1 (0.152 g, 0.137 mmol) in THF (4 mL). Pinkish-red solid 4, Yield: 0.162 g, 87%. Anal. Calcd for C36H20I2F6N6Pt2S2: C, 31.82; H, 1.48; N, 6.19; Found: C, 31.58; H, 1.32; N, 6.28. 1H-NMR (CD2Cl2, 400.16 MHz, 298K): δ 10.34 (2H, d, 3J(H6',H5') = 6.0; H6'), 8.28 (2H, dd, 3J(H2,H3) = 5.4, 4J(H2,H4) = 1.1, H2), 8.05 (2H, dd, 3J(H4,H3) = 8.0, 4J(H4,H2) = 1.1, H4), 7.57 (2H, dd, H3), 7.35 (2H, d, H5'), 7.30 (4H, AB, 3J(H5,H6) = 8.7 Hz, H5, H6), 6.93 (2H, dd, 3J(H9,H8) = 7.5; 4J(H9,H7) = 0.8, 3J(Pt,H9) = 31.4, H9), 6.86 (2H, d, 3J(H7,H8) = 7.8; H7), 6.71 (2H, dd, H8) ppm; 19F-NMR (CD2Cl2, 376 MHz, 298K): δ-70.6 (s, CF3) ppm; IR (cm−1): 1620 d, 1585 m, 1574 m, 1546 m, 1453 m, 1436 m, 1407 m, 1332 mf, 1322 mf, 1205 f, 1187 m, 1158 m, 1138 f, 1116 mf, 928 m, 845 m, 829 mf, 819 m, 763 f, 731 f, 709 f, 673 f, 653 m, 512 m, 482m, 467 m, 438 m. (Method B) A solution of HI in H2O 1M (292 µL, 0.292 mmol) was added to a pinkish-red suspension of 1 (0.147 g, 0.133 mmol) in THF (10 mL) and the mixture was stirred for 26 h in absence of light. During this time, a reddish-garnet solid was precipitated. The solvent was evaporated to ~2 mL and 10 mL of Et2O was added. The reddish-garnet solid 4·THF was filtered-off, washed with Et2O (2 × 5 mL) and dried. Yield: 0.156 g, 82%.
X-ray Structure Determinations. Crystal data and other details of the structure analyses are presented in
Table 2. Suitable crystals for X-ray diffraction studies were obtained by slow diffusion of
n-hexane into concentrated solutions of the complexes in 3 mL of CH
2Cl
2. Crystals were mounted at the end of quartz fibres. The radiation used in both cases was graphite monochromated MoKα (λ = 0.71073 Å). X-ray intensity data were collected on an Oxford Diffraction Xcalibur diffractometer. The diffraction frames were integrated and corrected from absorption using the CrysAlis RED program [
34]. The structures were solved by Patterson and Fourier methods and refined by full-matrix least squares on
F2 with SHELXL-97 [
35]. All non-hydrogen atoms were assigned anisotropic displacement parameters and refined without positional constraints. All hydrogen atoms were constrained to idealized geometries and assigned isotropic displacement parameters equal to 1.2 times the
Uiso values of their attached parent atoms. Full-matrix least-squares refinement of these models against
F2 converged to final residual indices given in
Table 2. CCDC 1011979–1011980 contain the supplementary crystallographic data for compounds
2·CH
2Cl
2 and
3·CH
2Cl
2[
36].
Table 2.
Crystal data and structure refinement for complexes [{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)Cl}2]·CH2Cl2 (2·CH2Cl2), [{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)Br}2]·CH2Cl2 (3·CH2Cl2).
Table 2.
Crystal data and structure refinement for complexes [{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)Cl}2]·CH2Cl2 (2·CH2Cl2), [{Pt(bzq)(µ-(CF3)C4H2N2S-κN,S)Br}2]·CH2Cl2 (3·CH2Cl2).
| Parameters | 2·CH2Cl2 | 3·CH2Cl2 |
|---|
| C37H22Cl4F6N6Pt2S2 | C37H22Br2Cl2F6N6Pt2S2 |
|---|
| Mt [g mol−1] | 1260.71 | 1349.63 |
| T [K] | 100(2) | 100(2) |
| λ [Å] | 0.71073 | 0.71073 |
| crystal system | triclinic | triclinic |
| space group | P-1 | P-1 |
| a [Å] | 11.3613(3) | 11.4798(2) |
| b [Å] | 11.8938(3) | 11.8668(2) |
| c [Å] | 14.2579(3) | 14.4711(3) |
| α [°] | 90.003(2) | 88.942(2) |
| β [°] | 98.289(2) | 81.111(2) |
| γ [°] | 101.170(2) | 78.410(2) |
| V [Å3] | 1869.64(8) | 1907.83(6) |
| Z | 2 | 2 |
| ρ [g cm−3] | 2.239 | 2.349 |
| μ [mm−1] | 7.942 | 9.737 |
| F(000) | 1192 | 1264 |
| 2
θ range [°] | 4.21–28.88 | 4.19–28.88 |
| no. of reflns collected | 41040 | 41763 |
| no. of unique reflns | 8961 | 9166 |
| R(int) | 0.0302 | 0.0318 |
| final
R indices [I > 2θ(I)] a | | |
| R1 | 0.0207 | 0.0232 |
| wR2 | 0.0446 | 0.0516 |
| R indices (all data) | | |
| R1 | 0.0250 | 0.0277 |
| wR2 | 0.0465 | 0.0534 |
| Goodness-of-fit on
F2 b | 1.024 | 1.058 |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

