
New Lanthanide Alkynylamidinates and Diiminophosphinates


Abstract
:
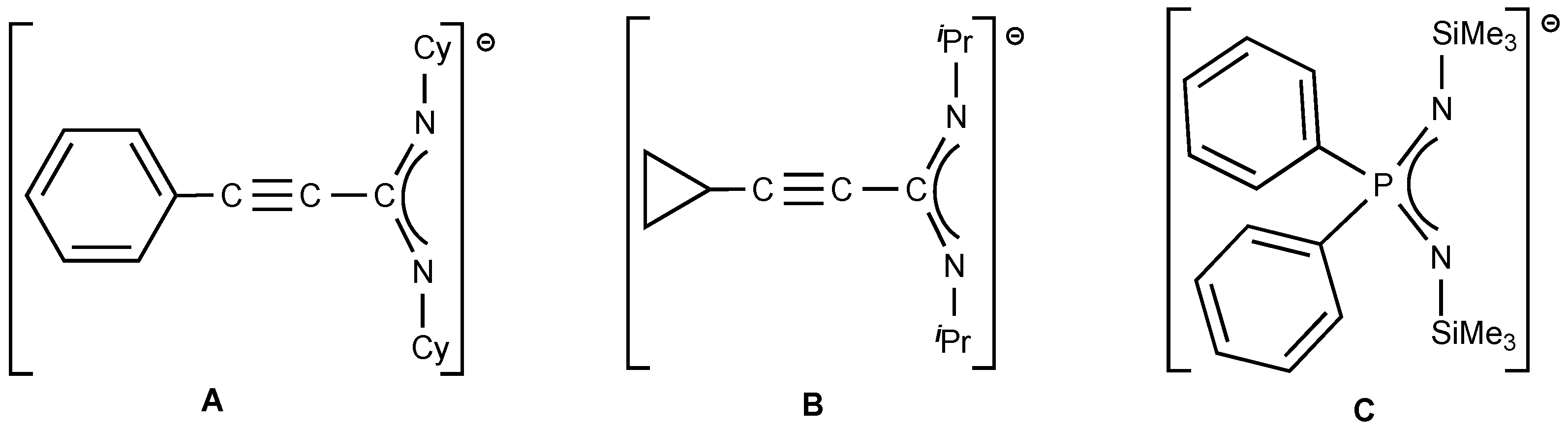
1. Introduction

2. Results and Discussion
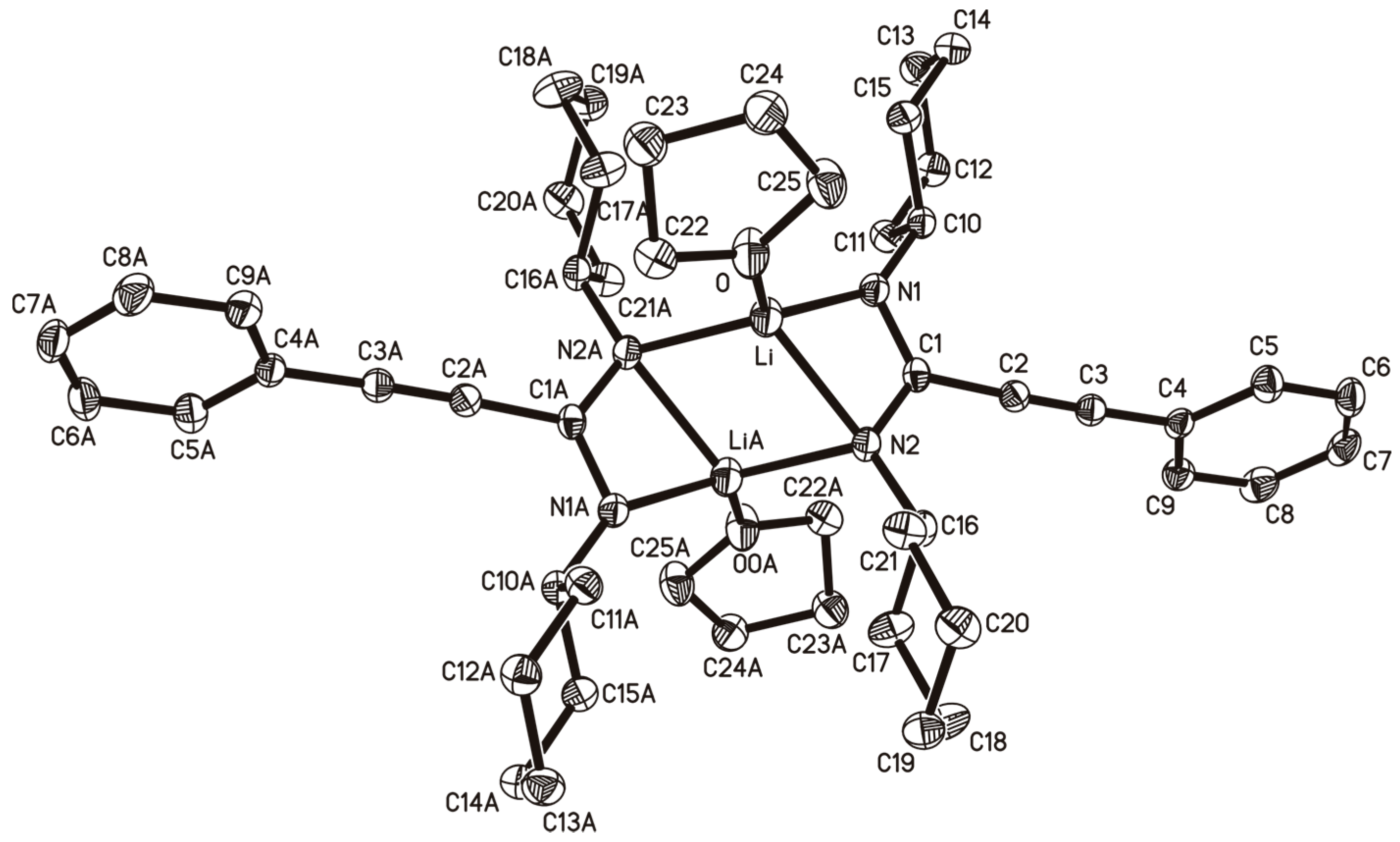
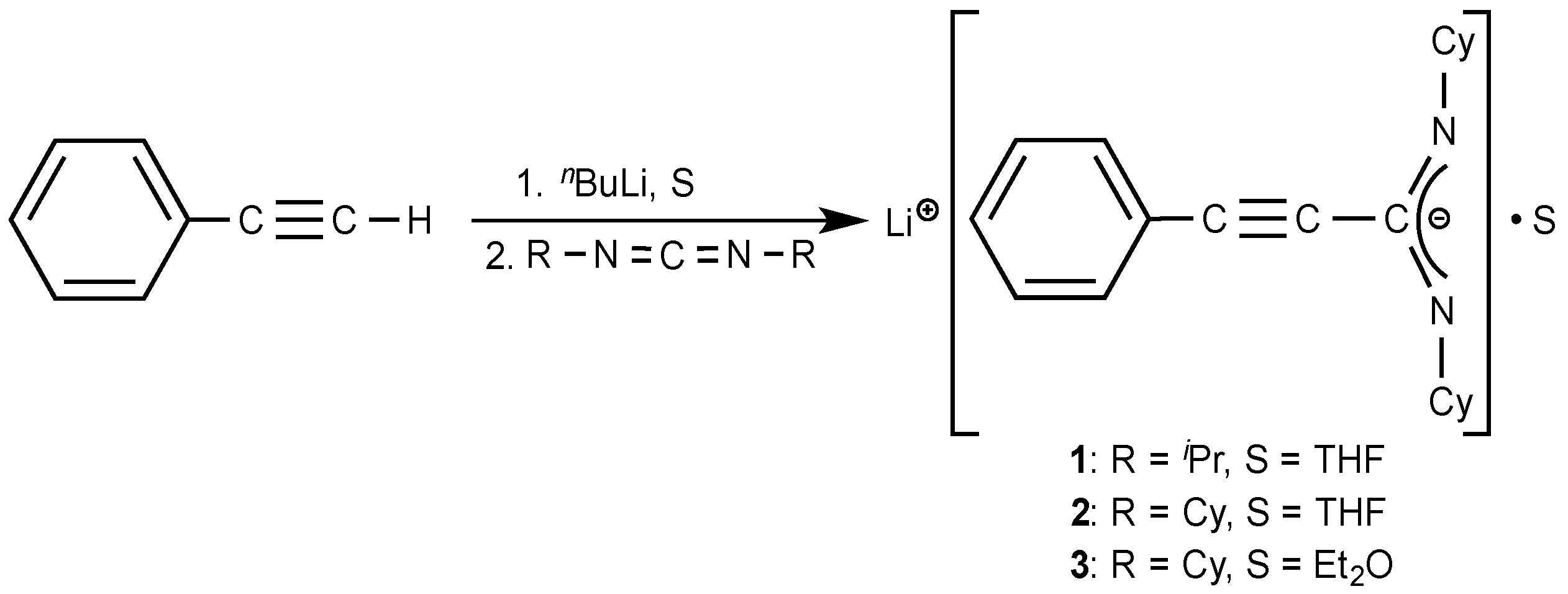
2.1. Lithium-Propiolamidinate Precursors



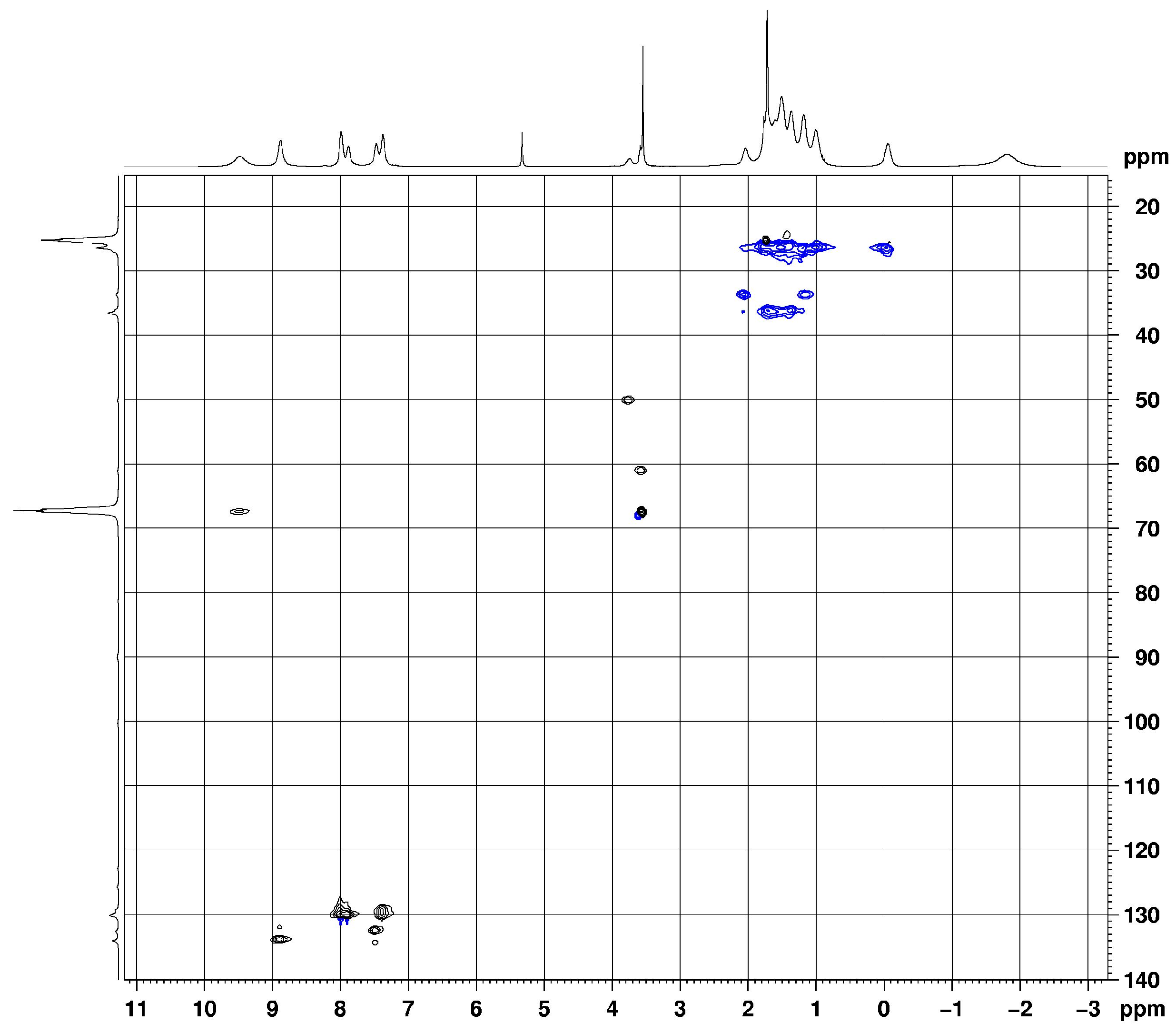
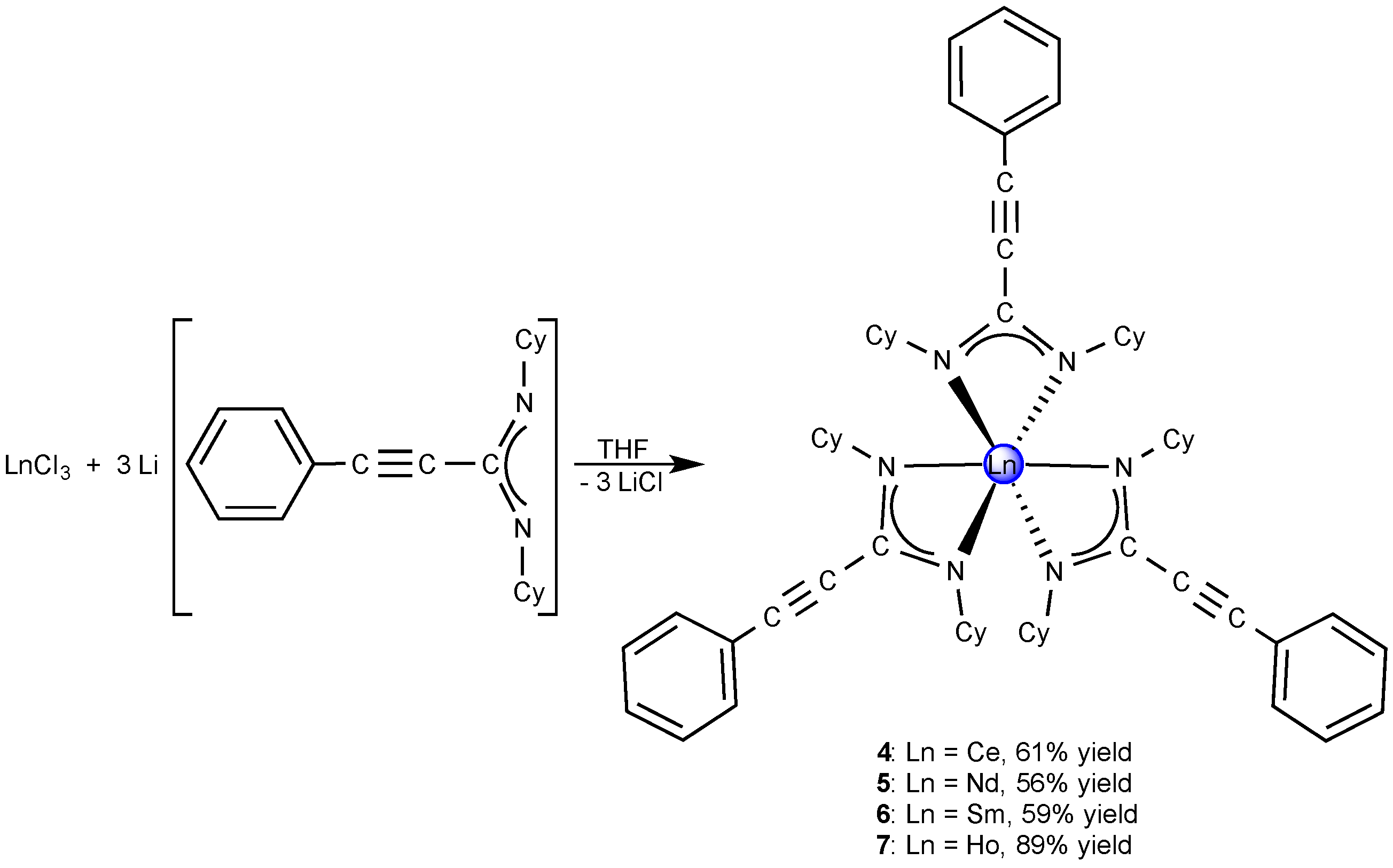
2.2. Homoleptic Lanthanide(III)-tris(Propiolamidinate) Complexes


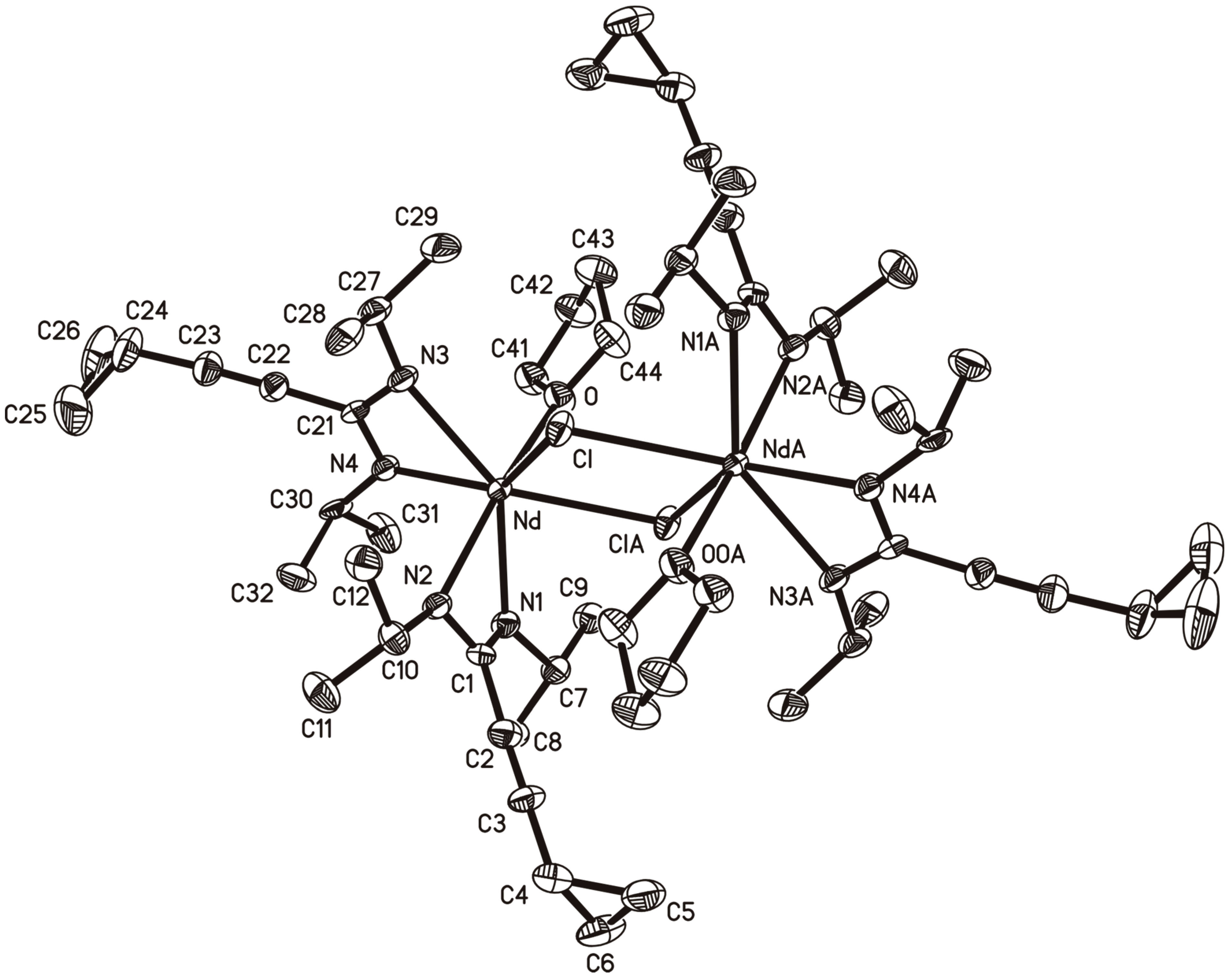
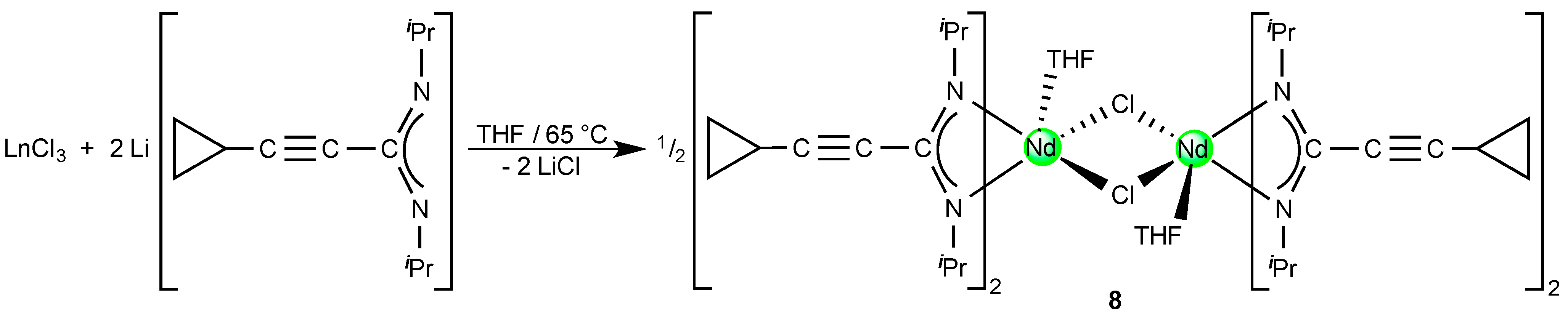
2.3. A Neodymium(III)-bis(Cyclopropylethinylamidinate)


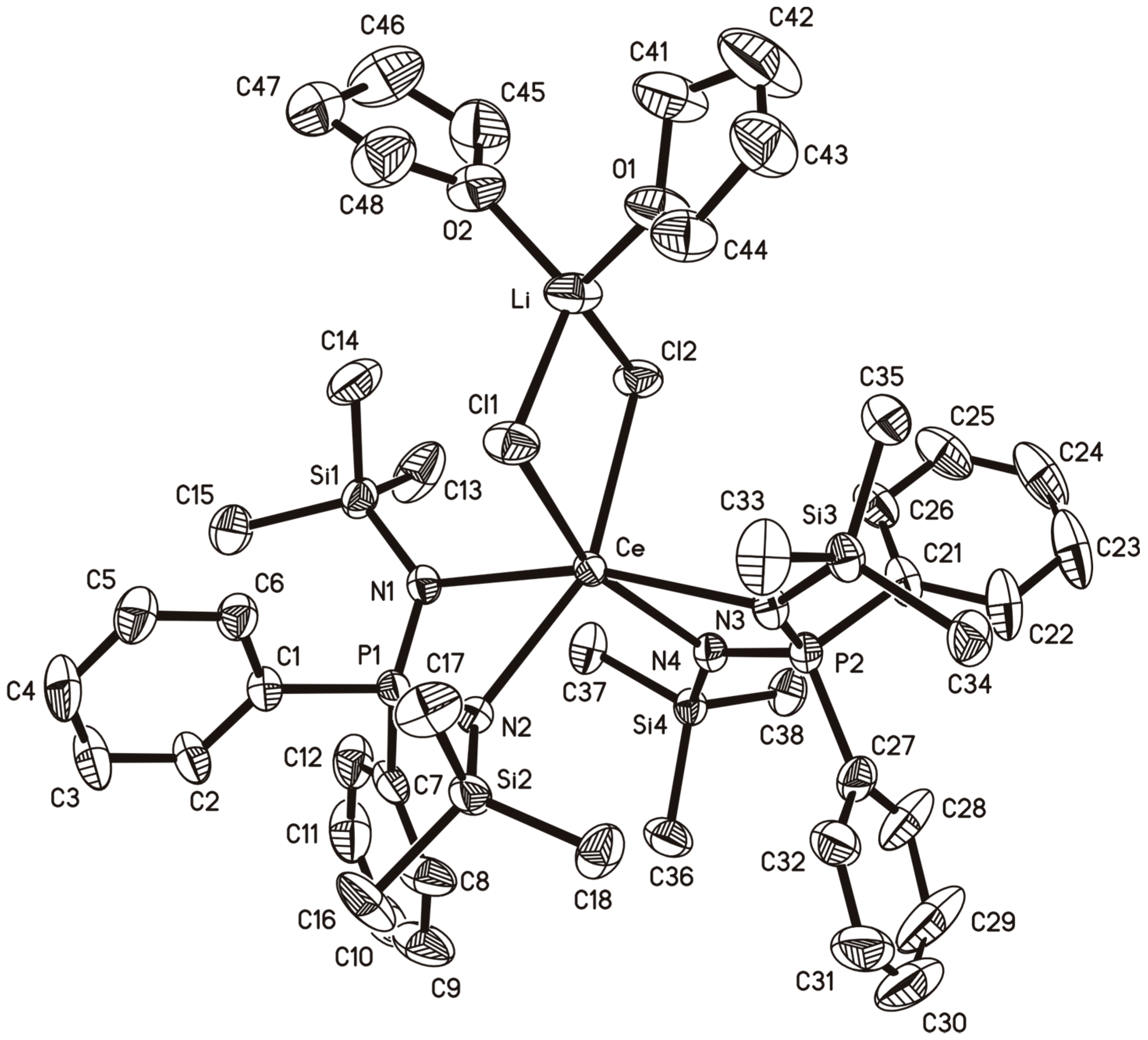
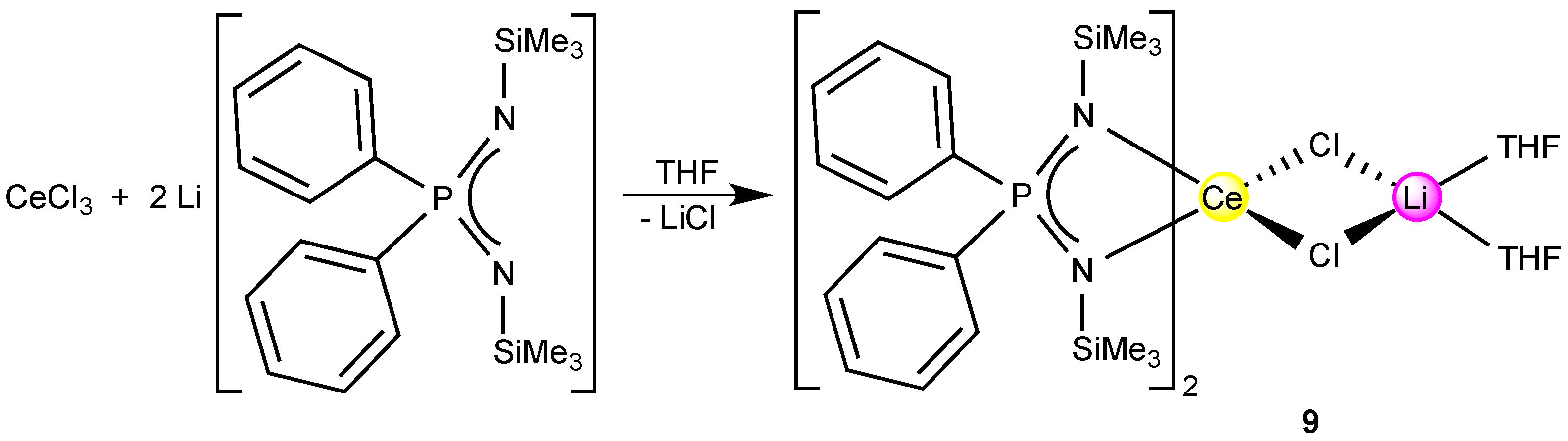
2.4. A Cerium(III)-bis(Diiminophosphinate) Complex


2.5. Attempted Oxidation of 9 to A Cerium(IV) Complex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1 | 2 | 8 | 9 |
|---|---|---|---|---|
| Empirical formula | C38H54Li2N4O2 | C50H70Li2N4O2 | C56H92Cl2N8Nd2O2 | C44H72CeCl2LiN4O2P2Si4 |
| a(Å) | 9.6687(19) | 9.8768(3) | 10.049(2) | 11.117(2) |
| b(Å) | 9.948(2) | 10.2082(3) | 11.337(2) | 36.425(7) |
| c(Å) | 19.611(4) | 11.6883(4) | 14.471(3) | 13.538(3) |
| α(°) | 93.77(3) | 104.144(3) | 82.53(3) | 90 |
| β(°) | 91.33(3) | 90.248(3) | 83.02(3) | 96.70(3) |
| γ(°) | 90.96(3) | 104.209(2) | 73.74(3) | 90 |
| V(Å3) | 1881.3(6) | 1105.18(6) | 1562.9(5) | 5444.3(19) |
| Z | 2 | 1 | 1 | 4 |
| formula weight | 612.73 | 772.98 | 1268.76 | 1081.32 |
| space group | P-1 | P-1 | P-1 | P21/n |
| T (°C) | −120 | −173 | −120 | −120 |
| λ (Å) | 0.71073 | 1.54184 | 0.71073 | 0.71073 |
| Dcalcd (g cm−3) | 1.082 | 1.161 | 1.348 | 1.319 |
| μ (mm−1) | 0.066 | 0.530 | 1.771 | 1.118 |
| F(000) | 664 | 420 | 654 | 2244 |
| data/restraints/parameters | 7395/0/469 | 4628/0/262 | 8340/0/324 | 12,841/0/569 |
| goodness-of-fit on F2 | 1.013 | 1.027 | 1.219 | 0.977 |
| R (Fo or Fo2) | 0.0520 | 0.0392 | 0.0547 | 0.0363 |
| Rw (Fo or Fo2) | 0.1347 | 0.0974 | 0.1663 | 0.0859 |
3. Experimental Section
3.1. General Procedures
3.2. Syntheses
3.3. X-ray Crystallography
4. Conclusions
Supplementary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Edelmann, F.T. Advances in the coordination chemistry of amidinate and guanidinate ligands. Adv. Organomet. Chem. 2008, 57, 183–352. [Google Scholar]
- Coles, M.P. Bicyclic-guanidines, -guanidinates and -guanidinium salts: Wide ranging applications from a simple family of molecules. Chem. Commun. 2009, 3659–3676. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Bulky guanidinates for the stabilization of low oxidation state metallacycles. Coord. Chem. Rev. 2010, 254, 1273–1289. [Google Scholar] [CrossRef]
- Trifonov, A.A. Guanidinate and amidopyridinate rare-earth complexes: Towards highly reactive alkyl and hydrido species. Coord. Chem. Rev. 2010, 254, 1327–1347. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Abdou, H.E., Jr.; Fackler, J.P. Coordination chemistry of gold(II) with amidinate, thiolate and ylide ligands. Coord. Chem. Rev. 2010, 254, 1253–1259. [Google Scholar] [CrossRef]
- Collins, S. Polymerization catalysis with transition metal amidinate and related complexes. Coord. Chem. Rev. 2011, 255, 118–138. [Google Scholar] [CrossRef]
- Edelmann, F.T. Recent progress in the chemistry of metal amidinates and guanidinates: Syntheses, catalysis and materials. Adv. Organomet. Chem. 2013, 61, 55–374. [Google Scholar]
- Edelmann, F.T. Lanthanide Amidinates and guanidinates: From laboratory curiosities to efficient homogeneous catalysts and precursors for rare-earth oxide thin films. Chem. Soc. Rev. 2009, 38, 2253–2268. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, F.T. Lanthanide amidinates and guanidinates in catalysis and materials science: A continuing success story. Chem. Soc. Rev. 2012, 41, 7657–7672. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Endo, R.; Aoyama, A.; Ichii, T. Propiolamidines I. syntheses of N,N′-disubstituted phenylpropiolamidines and new routes to 5-N-substituted amino-3-phenylisoxazoles and 5-N-substituted amino-1,3-diphenylpyrazoles. Bull. Chem. Soc. Jpn. 1972, 45, 1846–1852. [Google Scholar] [CrossRef]
- Himbert, G.; Feustel, M.; Jung, M. Aminoethinylierungen, 2. Synthese von (Aminoethinyl)ketonen über β-stannylierte Inamine. Liebigs Ann. Chem. 1981, 1907–1927. [Google Scholar] [CrossRef]
- Himbert, G.; Schwickerath, W. (Aminoethinyl)metallierungen, 13. Synthese und reaktionen von 3-aminopropiolamidinen. Liebigs Ann. Chem. 1984, 85–97. [Google Scholar] [CrossRef]
- Schmidt, G.F.; Süss-Fink, G. Katalytische C–C– und C–N–kupplungsreaktionen von carbodiimiden durch übergangsmetallcluster. J. Organomet. Chem. 1988, 356, 207–211. [Google Scholar] [CrossRef]
- Ong, T.-G.; O’Brien, J.S.; Korobkov, I.; Richeson, D.S. Facile and atom-efficient amidolithium-catalyzed C–C and C–N formation for the construction of substituted guanidines and propiolamidines. Organometallics 2006, 25, 4728–4730. [Google Scholar] [CrossRef]
- Xu, X.; Gao, J.; Cheng, D.; Li, J.; Qiang, G.; Guo, H. Copper-catalyzed highly efficient multicomponent reactions of terminal alkynes, acid chlorides, and carbodiimides: Synthesis of functionalized propiolamidine derivatives. Adv. Synth. Catal. 2008, 350, 61–64. [Google Scholar] [CrossRef]
- Dröse, P.; Hrib, C.G.; Edelmann, F.T. Synthesis and structural characterization of a homoleptic cerium(III) propiolamidinate. J. Organomet. Chem. 2010, 695, 1953–1956. [Google Scholar] [CrossRef]
- Weingärtner, W.; Kantlehner, W.; Maas, G. A convenient synthesis and some characteristic reactions of novel propiolamidinium salts. Synthesis 2011, 42, 265–272. [Google Scholar]
- Weingärtner, W.; Maas, G. Cycloaddition reactions of propiolamidinium salts. Eur. J. Org. Chem. 2012, 2012, 6372–6382. [Google Scholar] [CrossRef]
- Devi, A. “Old Chemistries” for new applications: Perspectives for development of precursors for MOCVD and ALD applications. Coord. Chem. Rev. 2013, 257, 3332–3384. [Google Scholar] [CrossRef]
- Fujita, H.; Endo, R.; Murayama, K.; Ichii, T. A novel cleavage of carbon–carbon triple bond. Bull. Chem. Soc. Jpn. 1972, 45, 1581. [Google Scholar] [CrossRef]
- Ried, W.; Wegwitz, M. Synthese von speziell substituierten 1,3-Dithiol-2-thionen und 1,3-Thiaselenol-2-thionen. Liebigs Ann. Chem. 1975, 1975, 89–94. [Google Scholar] [CrossRef]
- Ried, W.; Schweitzer, R. Neue cyclisierungsprodukte aus N,N′-diarylsubstituierten propiolamidinen. Chem. Ber. 1976, 109, 1643–1649. [Google Scholar] [CrossRef]
- Ried, W.; Winkler, H. Cyclisierungsprodukte aus N-arylsubstituierten propiolamidinen. Chem. Ber. 1979, 112, 384–388. [Google Scholar] [CrossRef]
- Sienkiewich, P.; Bielawski, K.; Bielawska, A.; Palka, J. Inhibition of collagen and DNA biosynthesis by a novel amidine analogue of chlorambucil is accompanied by deregulation of β1-integrin and IGF-I receptor signaling in MDA-MB 231 cells. Environ. Toxicol. Pharmacol. 2005, 20, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Sielecki, T.M.; Liu, J.; Mousa, S.A.; Racanelli, A.L.; Hausner, E.A.; Wexler, R.R.; Olson, R.E. Synthesis and pharmacology of modified amidine isoxazoline glycoprotein IIb/IIIa receptor antagonists. Bioorg. Med. Chem. Lett. 2001, 11, 2201–2204. [Google Scholar] [CrossRef]
- Stephens, C.E.; Tanious, E.; Kim, S.; Wilson, D.W.; Schell, W.A.; Perfect, J.R.; Franzblau, S.G.; Boykin, D.W. Diguanidino and “reversed” diamidino 2,5-diarylfurans as antimicrobial agents. J. Med. Chem. 2001, 44, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Rowley, C.N.; DiLabio, G.A.; Barry, S.T. Theoretical and synthetic investigations of carbodiimide insertions into Al–CH3 and Al–N(CH3)2 bonds. Inorg. Chem. 2005, 44, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-X.; Nishiura, M.; Hou, Z. Catalytic addition of terminal alkynes to carbodiimides by half-sandwich rare earth metal complexes. J. Am. Chem. Soc. 2005, 127, 16788–16789. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, S.; Yang, G.; Li, Q.; Zhang, L.; Yao, Z.; Zhou, Z.; Song, H.-B. Synthesis, structure, and diverse catalytic activities of [ethylenebis(indenyl)]lanthanide(III) amides on N–H and C–H addition to carbodiimides and ε-caprolactone polymerization. Organometallics 2007, 26, 3755–3761. [Google Scholar] [CrossRef]
- Zhang, W.-X.; Hou, Z. Catalytic addition of alkyne C–H, amine N–H, and phosphine P–H bonds to carbodiimides: An efficient route to propiolamidines, guanidines, and phosphaguanidines. Org. Biomol. Chem. 2008, 6, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Li, W.; Zhu, X.; Xu, F.; Shen, Q. Divalent lanthanide complexes: Highly active precatalysts for the addition of N–H and C–H bonds to carbodiimides. J. Org. Chem. 2008, 73, 8966–8972. [Google Scholar] [CrossRef] [PubMed]
- Rowley, C.N.; Ong, T.-G.; Priem, J.; Richeson, D.S.; Woo, T.K. Analysis of the critical step in catalytic carbodiimide transformation: Proton transfer from amines, phosphines, and alkynes to guanidinates, phosphaguanidinates, and propiolamidinates with Li and Al catalysts. Inorg. Chem. 2008, 47, 12024–12031. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, S.; Zhang, L.; Yang, G.; Zhu, X.; Zhou, Z.; Zhu, H.; Wu, S. Cyclopentadienyl-free rare-earth metal amides [{(CH2SiMe2){(2,6-iPr2C6H3)N}2}Ln{N(SiMe3)2}(THF)] as highly efficient versatile catalysts for C–C and C–N bond formation. Eur. J. Org. Chem. 2010, 41, 326–332. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.-C.; Zhang, W.-X.; Xi, Z. Half-sandwich bis(propiolamidinate) rare-earth metal complexes: Synthesis, structure and dissociation of the cyclopentadienyl ligand via competition with an amidinate. Dalton Trans. 2013, 42, 16466–16469. [Google Scholar] [CrossRef] [PubMed]
- Seidel, W.W.; Dachtler, W.; Pape, T. Synthesis, structure, and reactivity of RuII complexes with trimethylsilylethinylamidinate ligands. Z. Anorg. Allg. Chem. 2012, 638, 116–121. [Google Scholar] [CrossRef]
- Brown, D.J.; Chisholm, M.H.; Gallucci, J.C. Amidinate-carboxylate complexes of dimolybdenum and ditungsten: M2(O2CR)2((NiPr)2CR′)2. Preparations, molecular and electronic structures and reactions. Dalton Trans. 2008, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.; Kurtz, W.; Effenberger, F. NMR-spektroskopische untersuchung der konformation und reaktivität von cyclopropylbenzolen. Chem. Ber. 1973, 106, 549–559. [Google Scholar] [CrossRef]
- Wilcox, C.F.; Loew, L.M.; Hoffman, R. Why a cyclopropyl group is good at stabilizing a cation but poor at transmitting substituent effects. J. Am. Chem. Soc. 1973, 95, 8192–8193. [Google Scholar] [CrossRef]
- De Meijere, A. Bonding properties of cyclopropane and their chemical consequences. Angew. Chem. Int. Ed. 1979, 18, 809–826. [Google Scholar] [CrossRef]
- Brown, H.C.; Periasamy, M.; Perumal, P.T. Structural effects in solvolytic reactions. 47. Effects of p-alkyl and p-cycloalkyl groups on the carbon-13 NMR shifts of the cationic carbon center in p-alkyl-tert-cumyl cations. J. Org. Chem. 1984, 49, 2754–2757. [Google Scholar] [CrossRef]
- Roberts, D.D. Electron-donating ability of the cyclopropyl substituent in the solvolyses of an α-CF3-substituted secondary-alkyl tosylate. J. Org. Chem. 1991, 56, 5661–5665. [Google Scholar] [CrossRef]
- Sauers, R.R. Cyclopropanes: Calculation of NMR spectra by ab initio methodology. Tetrahedron 1998, 54, 337–348. [Google Scholar] [CrossRef]
- De Meijere, A.; Kozhushkov, S.I. Small rings in focus: Recent routes to cyclopropylgroup containing carbo- and heterocycles. Mendeleev Commun. 2010, 20, 301–311. [Google Scholar] [CrossRef]
- Sroor, F.M.A.; Hrib, C.G.; Hilfert, L.; Edelmann, F.T. Lithium-cyclopropylethinylamidinates. Z. Anorg. Allg. Chem. 2013, 639, 2390–2394. [Google Scholar] [CrossRef]
- Sroor, F.M.; Hrib, C.G.; Hilfert, L.; Jones, P.G.; Edelmann, F.T. Lanthanide(III)-bis(cyclopropylethinylamidinates): Synthesis, structure, and catalytic activity. J. Organomet. Chem. 2015, 785, 1–10. [Google Scholar] [CrossRef]
- Sroor, F.M.; Hrib, C.G.; Hilfert, L.; Busse, S.; Edelmann, F.T. Synthesis and catalytic activity of homoleptic lanthanide-tris(cyclopropylethinyl)amidinates. New J. Chem. 2015, 39, 7595–7601. [Google Scholar] [CrossRef]
- Sroor, F.M.; Hrib, C.G.; Hilfert, L.; Hartenstein, L.; Roesky, P.W.; Edelmann, F.T. Synthesis and structural characterization of new bis(alkynylamidinato)lanthanide(III)-amides. J. Organomet. Chem. 2015, 799–800, 160–165. [Google Scholar] [CrossRef]
- Stalke, D.; Wedler, M.; Edelmann, F.T. Dimere Alkalimetallbenzamidinate: Einfluß des metallions auf die struktur. J. Organomet. Chem. 1992, 431, C1–C5. [Google Scholar] [CrossRef]
- Barker, J.; Barr, D.; Barnett, N.D.R.; Clegg, W.; Cragg-Hine, I.; Davidson, M.G.; Davies, R.P.; Hodgson, S.M.; Howard, J.A.K.; Kilner, M.; et al. Lithiated amidines: Syntheses and structural characterisations. Dalton Trans. 1997, 951–955. [Google Scholar] [CrossRef]
- Hagadorn, J.R.; Arnold, J. Ferrocene-substituted amidinate derivatives: Syntheses and crystal structures of lithium, iron(II), and cobalt(II) complexes. Inorg. Chem. 1997, 36, 132–133. [Google Scholar] [CrossRef]
- Caro, C.F.; Hitchcock, P.B.; Lappert, M.F.; Layh, M. Neutral isonitrile adducts of alkali and alkaline earth metals. Chem. Commun. 1998, 1297–1298. [Google Scholar] [CrossRef]
- Knapp, C.; Lork, E.; Watson, P.G.; Mews, R. Lithium fluoroarylamidinates: Syntheses, structures, and reactions. Inorg. Chem. 2002, 41, 2014–2025. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.L.; Tyrrell, B.R.; Mountford, P. Bis(µ-N-n-propyl-N-trimethylsilylbenz-amidinato)bis[(diethyl ether-O)lithium. Acta Crystallogr. Sect. E 2002, 58, m597–m598. [Google Scholar] [CrossRef]
- Aharonovich, S.; Kapon, M.; Botoshanski, M.; Eisen, M.S. N,N′-bis-silylated lithium aryl amidinates: Synthesis, characterization, and the gradual transition of coordination mode from σ toward π originated by crystal packing interactions. Organometallics 2008, 27, 1869–1877. [Google Scholar] [CrossRef]
- Nevoralová, J.; Chlupaty, T.; Padelková, Z.; Ruzicka, A. Quest for lithium amidinates containing adjacent amino donor group at the central carbon atom. J. Organomet. Chem. 2013, 745–746, 186–189. [Google Scholar]
- Hong, J.; Zhang, L.; Wang, K.; Chen, Z.; Wu, L.; Zhou, X. Synthesis, structural characterization, and reactivity of mono(amidinate) rare-earth-metal bis(aminobenzyl) complexes. Organometallics 2013, 32, 7312–7322. [Google Scholar] [CrossRef]
- Snaith, R.; Wright, D.S. Lithium Chemistry, A Theoretical and Experimental Overview; Sapse, A., von R. Schleyer, P., Eds.; Wiley: New York, NY, USA, 1995. [Google Scholar]
- Downard, A.; Chivers, T. Applications of the laddering principle—A two-stage approach to describe lithium heterocarboxylates. Eur. J. Inorg. Chem. 2001, 2001, 2193–2221. [Google Scholar] [CrossRef]
- Richter, J.; Feiling, J.; Schmidt, H.-G.; Noltemeyer, M.; Brüser, W.; Edelmann, F.T. Lanthanide(III) amidinates with six- and seven-coordinate metal atoms. Z. Anorg. Allg. Chem. 2004, 630, 1269–1275. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, Y.; Chen, J.; Zhang, Z.; Zhang, Y.; Shen, Q. Synthesis and characterization of bis(guanidinate)lanthanide diisopropylamido complexes—New highly active initiators for the polymerizations of ε-caprolactone and methyl methacrylate. J. Organomet. Chem. 2003, 679, 229–237. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Bubnov, A.M.; Fukin, G.K.; Dolgushin, F.M.; Antipin, M.Y.; Pelcé, O.; Schappacher, M.; Guillaume, S.M.; Trifonov, A.A. Hydrido complexes of yttrium and lutetium supported by bulky guanidinato ligands [Ln(μ-H){(Me3Si)2NC(NCy)2}2]2 (Ln = Y, Lu): Synthesis, structure, and reactivity. Eur. J. Inorg. Chem. 2008, 2008, 2090–2098. [Google Scholar] [CrossRef]
- Recknagel, A.; Witt, M.; Edelmann, F.T. Synthese von metallacyclophosphazenen mit praseodym, neodym, uran und thorium. J. Organomet. Chem. 1989, 371, C40–C44. [Google Scholar] [CrossRef]
- Recknagel, A.; Steiner, A.; Noltemeyer, M.; Brooker, S.; Stalke, D.; Edelmann, F.T. Diiminophosphinate des lithiums, samariums und ytterbiums: Molekülstrukturen von Li[Ph2P(NSiMe3)2](THF)2 und [Ph2P(NSiMe3)2]2Sm(µ-I)2Li(THF)2. J. Organomet. Chem. 1991, 414, 327–335. [Google Scholar] [CrossRef]
- Schumann, H.; Winterfeld, J.; Hemling, H.; Hahn, F.E.; Reich, P.; Brzezinka, K.-W.; Edelmann, F.T.; Kilimann, U.; Schäfer, M.; Herbst-Irmer, R. (Cyclooctatetraenyl)[N,N′-bis(trimethylsilyl)benzamidinato]- und -[P,P-diphenyl-bis-(trimethylsilylimino)phosphinato]-Komplexe der Seltenen Erden: Röntgenstrukturanalyse von (C8H8)Tm[PhC(NSiMe3)2](THF), (C8H8)Lu[4-MeOC6H4C(NSiMe3)2](THF) und (C8H8)Nd[Ph2P(NSiMe3)2](THF). Chem. Ber. 1995, 128, 395–404. [Google Scholar]
- Liu, B.; Li, L.; Sun, G.; Liu, J.; Wang, M.; Li, S.; Cui, D. 3,4-Polymerization of Isoprene by using NSN- and NPN-ligated rare earth metal precursors: Switching of stereo selectivity and mechanism. Macromolecules 2014, 47, 4971–4978. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schwirten, K.; Pickel, H. Kleine anorganische ringe, II. ein cyclisches alumophosphazan und seine gallium- und Indiumanalogen. Chem. Ber. 1969, 102, 564–567. [Google Scholar] [CrossRef]
- Wolfsberger, W.; Hager, W. Phosphinimino- und arsinimino-phosphoniumhalogenide. Z. Anorg. Allg. Chem. 1977, 433, 247–254. [Google Scholar] [CrossRef]
- Edelmann, F.T. Comprehensive Organometallic Chemistry III, Vol. 3, Complexes of Scandium, Yttrium and Lanthanide Elements; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: Oxford, UK, 2006. [Google Scholar]
- Morton, C.; Alcock, N.W.; Lees, M.R.; Munslow, I.J.; Sanders, C.J.; Scott, P. Stabilization of cerium(IV) in the presence of an iodide ligand: Remarkable effects of lewis acidity on valence state. J. Am. Chem. Soc. 1992, 121, 11255–11256. [Google Scholar] [CrossRef]
- Eisenstein, O.; Hitchcock, P.B.; Hulkes, A.G.; Lappert, M.F.; Maron, L. Cerium masquerading as a group 4 element: Synthesis, structure and computational characterisation of [CeCl{N(SiMe3)2}3]. Chem. Commun. 2001, 17, 1560–1561. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Hulkes, A.G.; Lappert, M.F. Oxidation in nonclassical organolanthanide chemistry: Synthesis, characterization, and X-ray crystal structures of cerium(III) and -(IV) amides. Inorg. Chem. 2003, 43, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Werner, D.; Deacon, G.B.; Junk, P.C.; Anwander, R. Cerium(III/IV) formamidinate chemistry, and a stable cerium(IV) diolate. Chem. Eur. J. 2014, 20, 4426–4438. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Spallek, T.; Maichle-Mössmer, C.; Törnroos, K.W.; Anwander, R. Cerium tetrakis(diisopropylamide)—A useful precursor for cerium(IV) chemistry. Chem. Commun. 2014, 50, 14763–14766. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, P.B.; Lappert, M.F.; Protchenko, A.V. Facile formation of a homoleptic Ce(IV) amide via aerobic oxidation. Chem. Commun. 2006, 3546–3548. [Google Scholar] [CrossRef] [PubMed]
- Crozier, A.R.; Bienfait, A.M.; Maichle-Mössmer, C.; Törnroos, K.W.; Anwander, R. A homoleptic tetravalent cerium silylamide. Chem. Commun. 2013, 49, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.H.; Smith, M.L. The preparation of anhydrous inorganic chlorides by dehydration with thionyl chloride. J. Inorg. Nucl. Chem. 1958, 7, 224–227. [Google Scholar] [CrossRef]
- XAREA program for X-ray crystal data collection; XRED32; Stoe: Darmstadt, Germany, 2002.
- SMART, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 1998.
- Sheldrick, G.M. SHELXS-97 Program for Crystal Structure Solution; Universität Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97 Program for Crystal Structure Refinement; Universität Göttingen: Göttingen, Germany, 1997. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sroor, F.M.; Hrib, C.G.; Edelmann, F.T. New Lanthanide Alkynylamidinates and Diiminophosphinates. Inorganics 2015, 3, 429-447. https://doi.org/10.3390/inorganics3040429
Sroor FM, Hrib CG, Edelmann FT. New Lanthanide Alkynylamidinates and Diiminophosphinates. Inorganics. 2015; 3(4):429-447. https://doi.org/10.3390/inorganics3040429
Chicago/Turabian StyleSroor, Farid M., Cristian G. Hrib, and Frank T. Edelmann. 2015. "New Lanthanide Alkynylamidinates and Diiminophosphinates" Inorganics 3, no. 4: 429-447. https://doi.org/10.3390/inorganics3040429
APA StyleSroor, F. M., Hrib, C. G., & Edelmann, F. T. (2015). New Lanthanide Alkynylamidinates and Diiminophosphinates. Inorganics, 3(4), 429-447. https://doi.org/10.3390/inorganics3040429