
First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
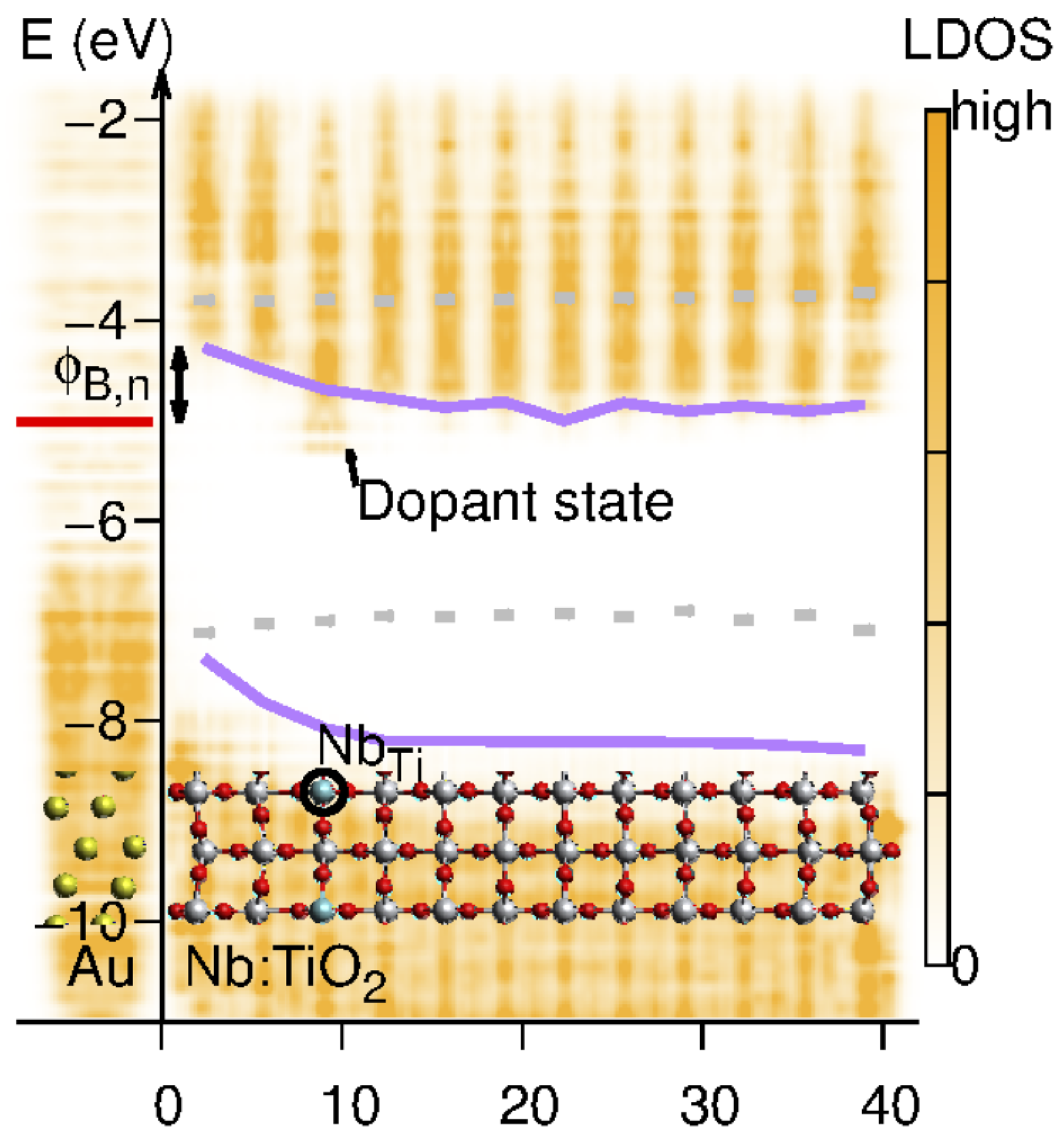{kind=link}
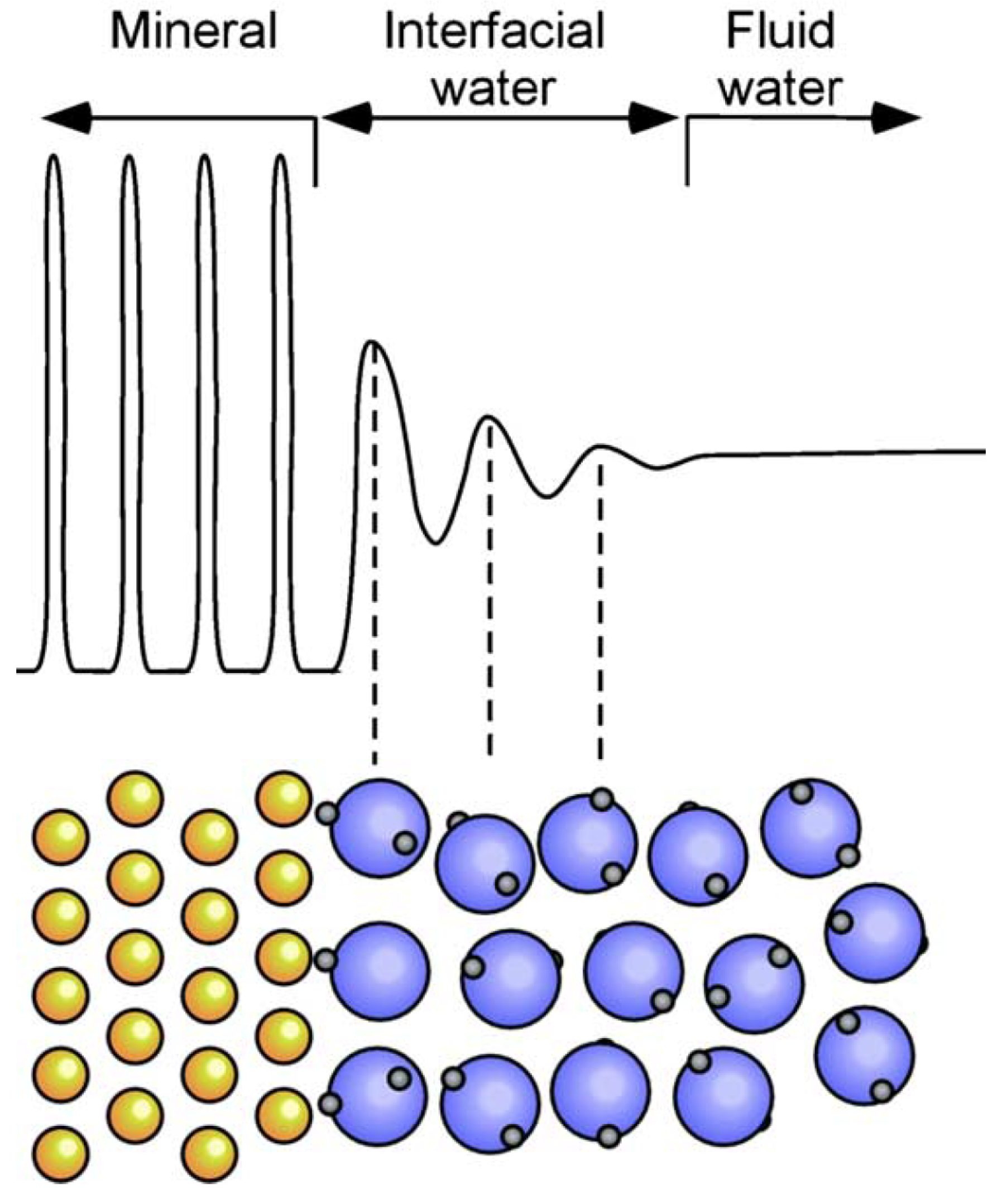{kind=link}
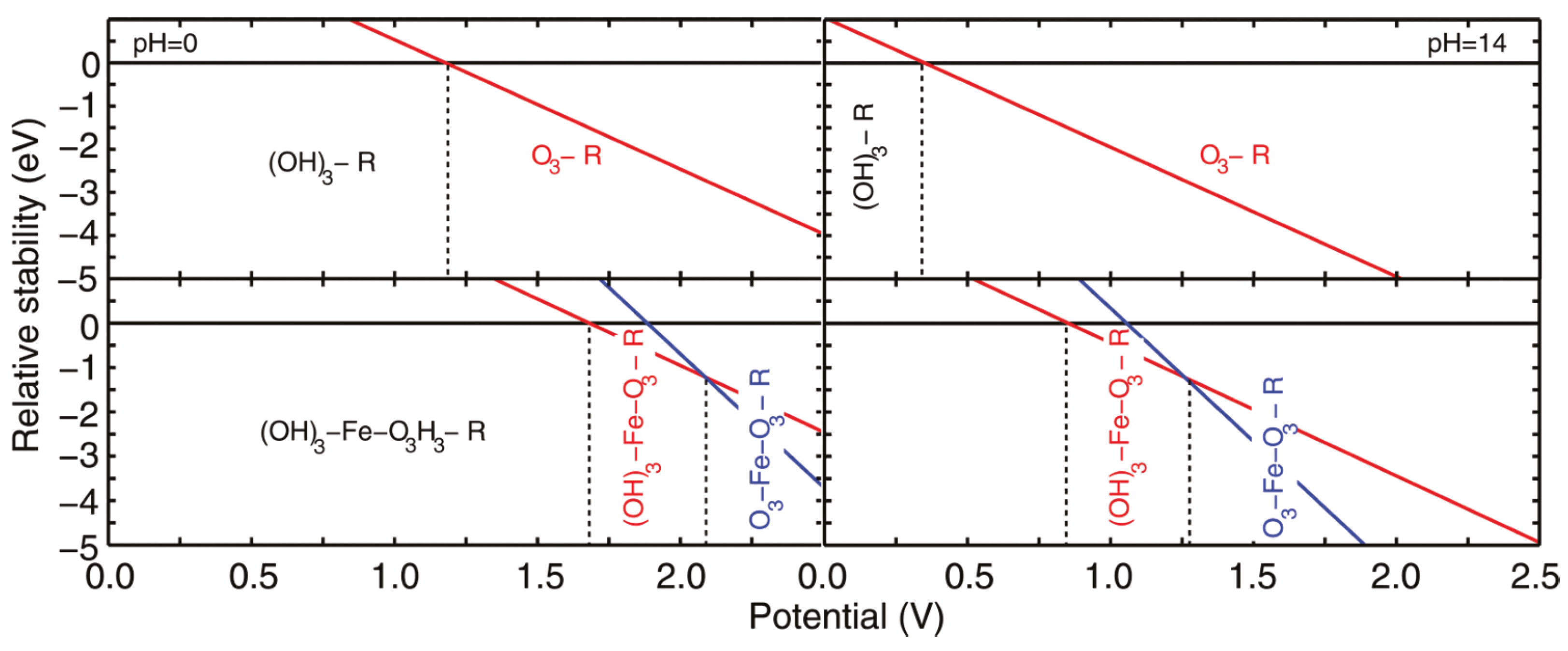{kind=link}
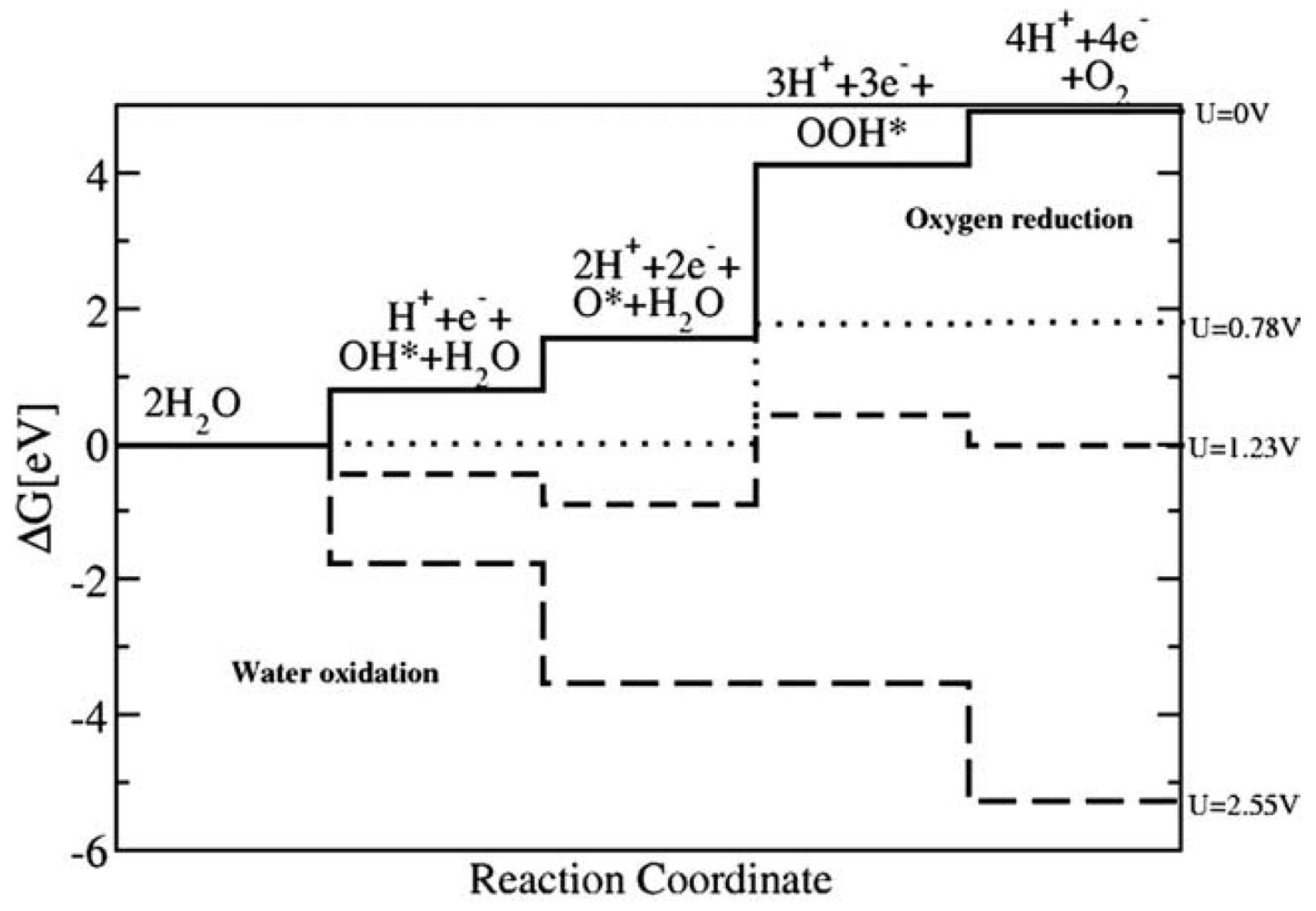{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
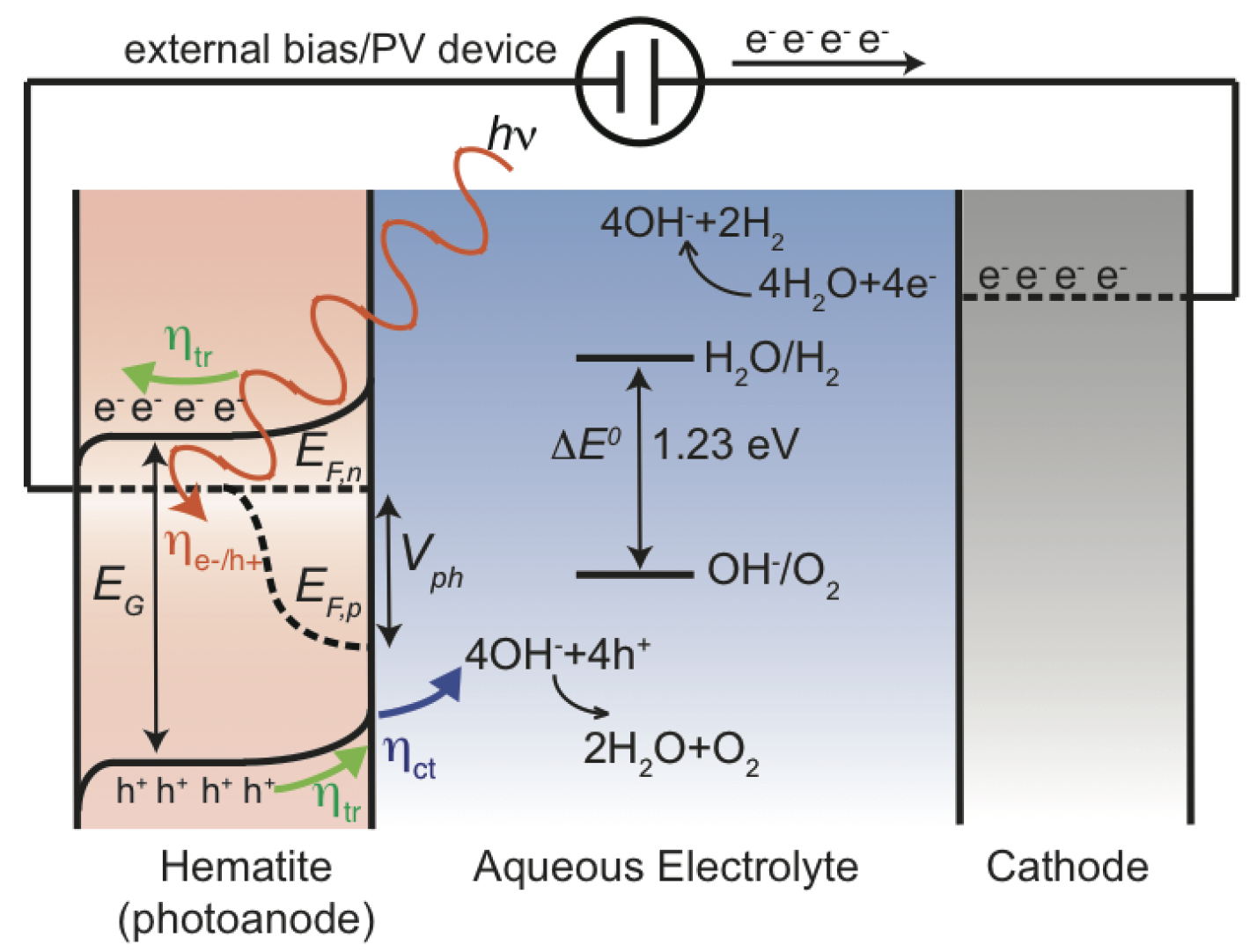
2. Processes Involved in Photoelectrochemistry
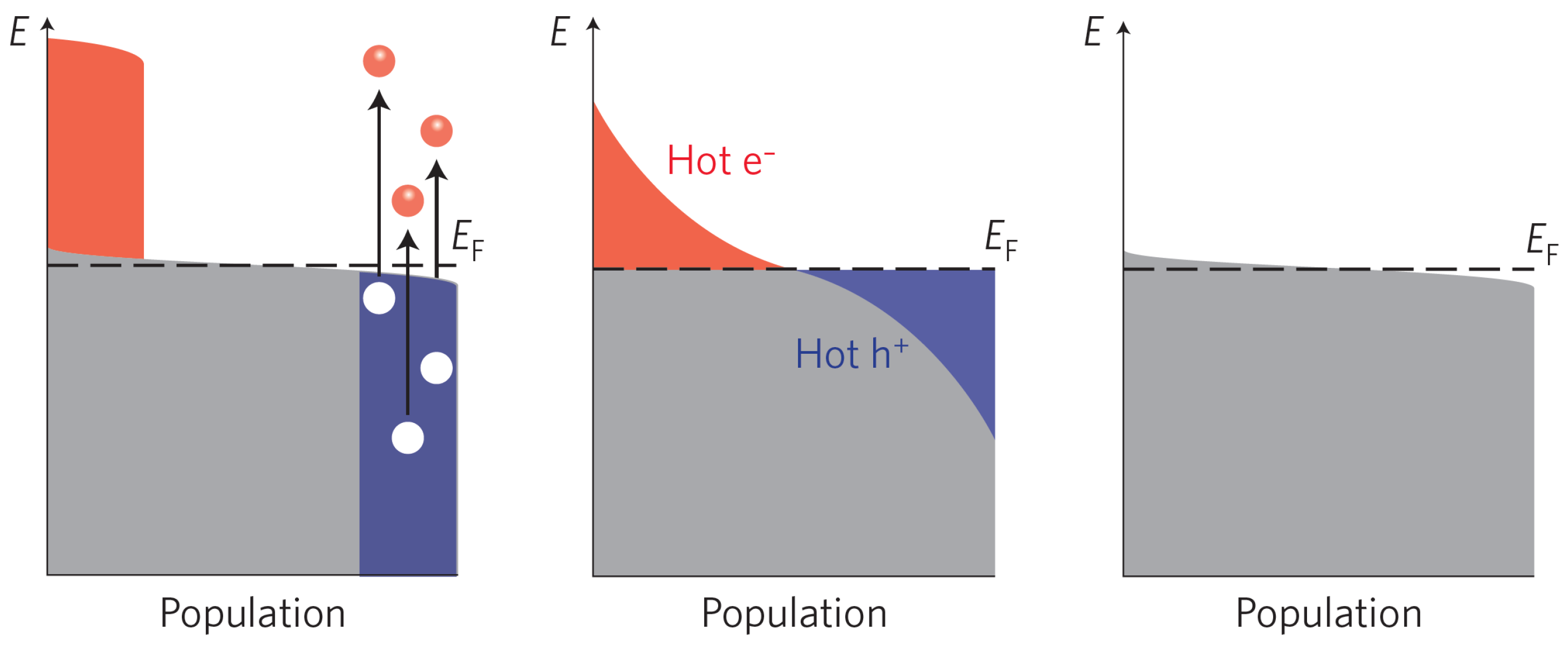
2.1. Creation of Electron-Hole Pairs
2.2. Charge Carrier Transport
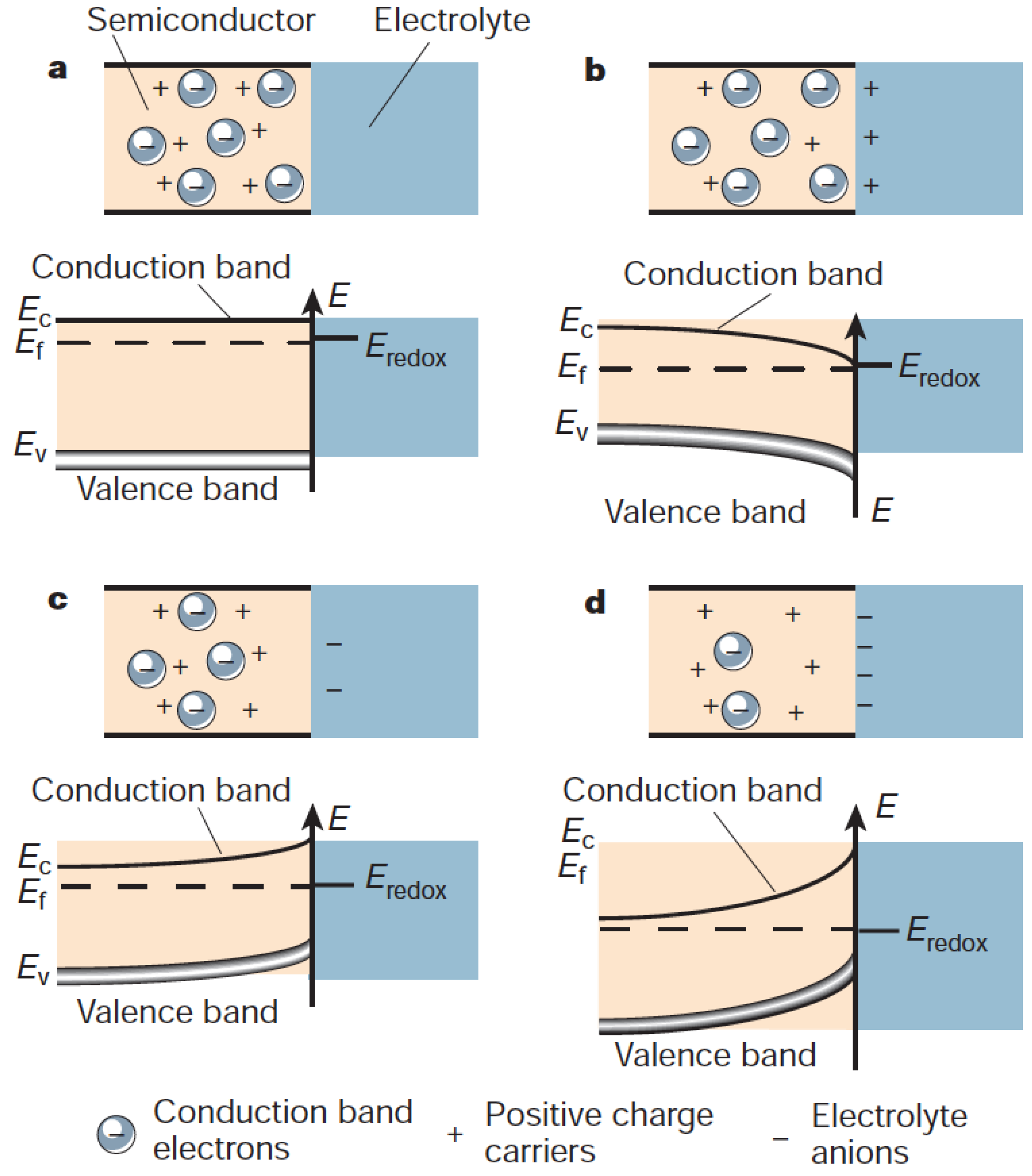
2.3. Electrochemical Surface Reactions
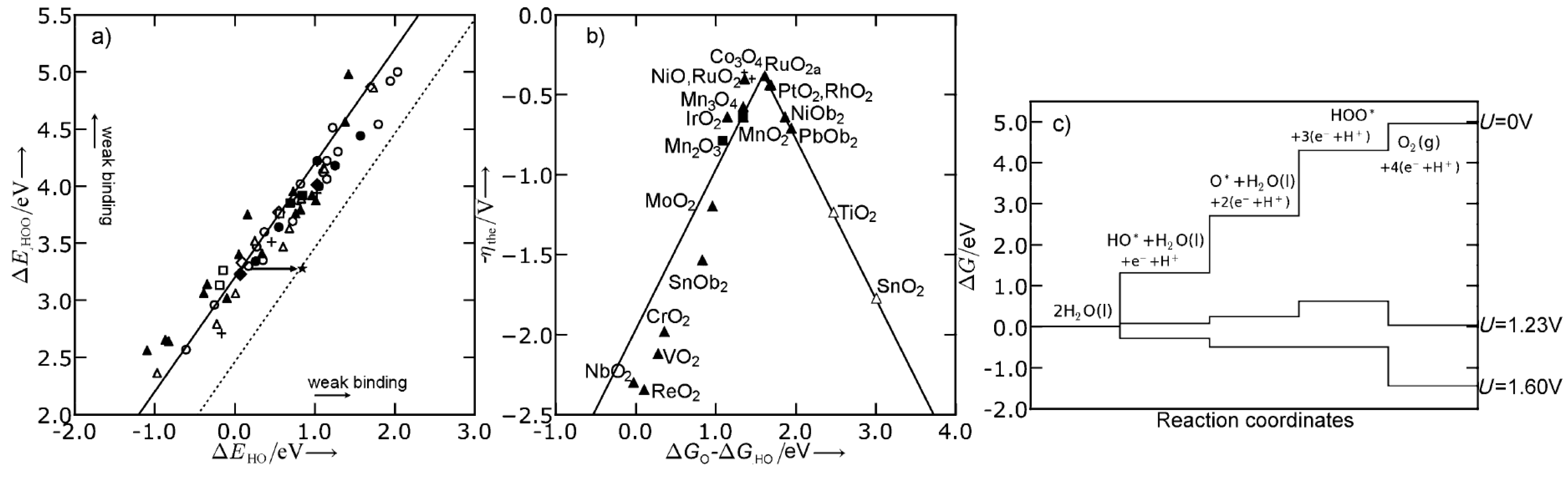
Pourbaix Surface Diagrams
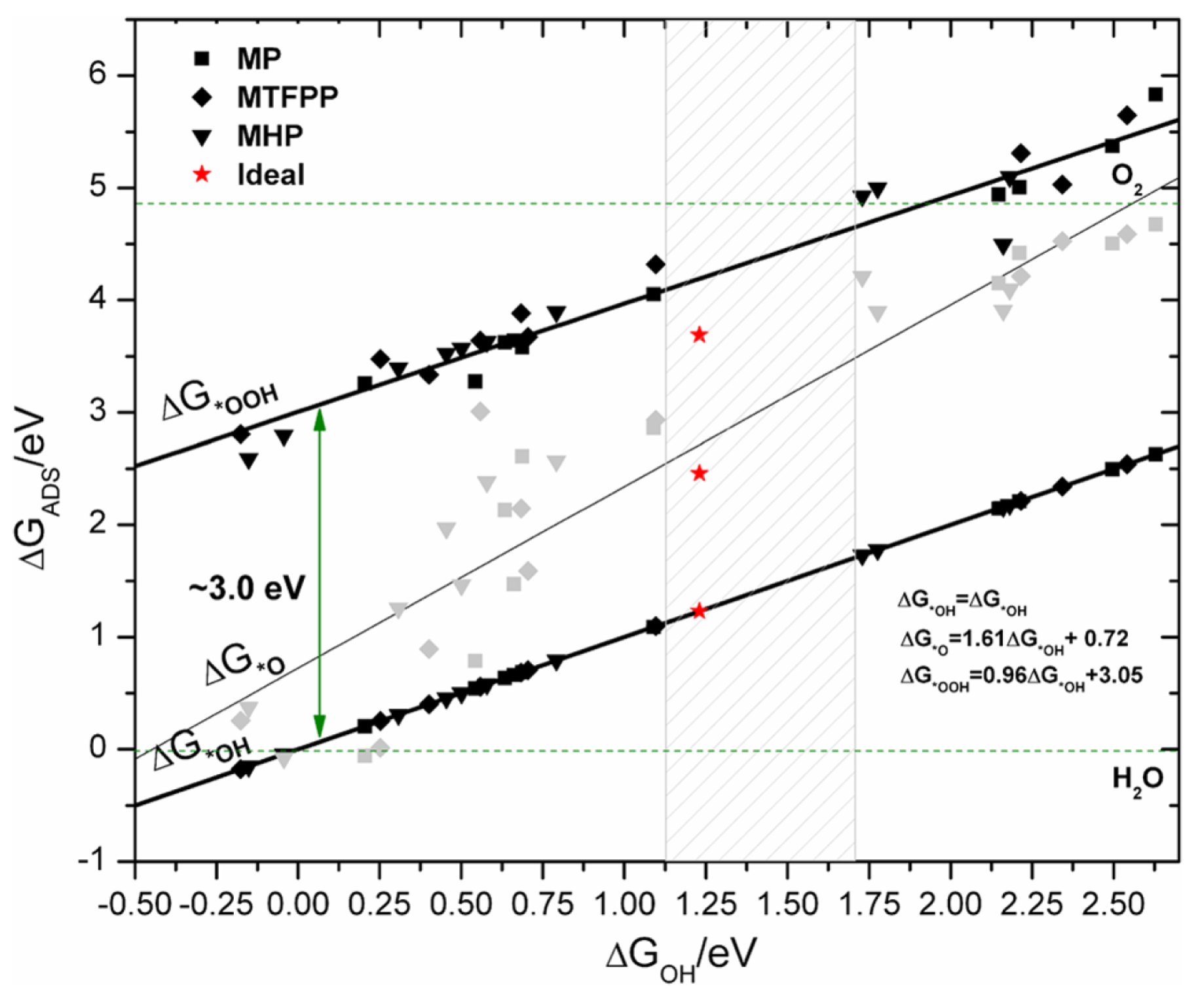
2.4. Reaction Mechanism
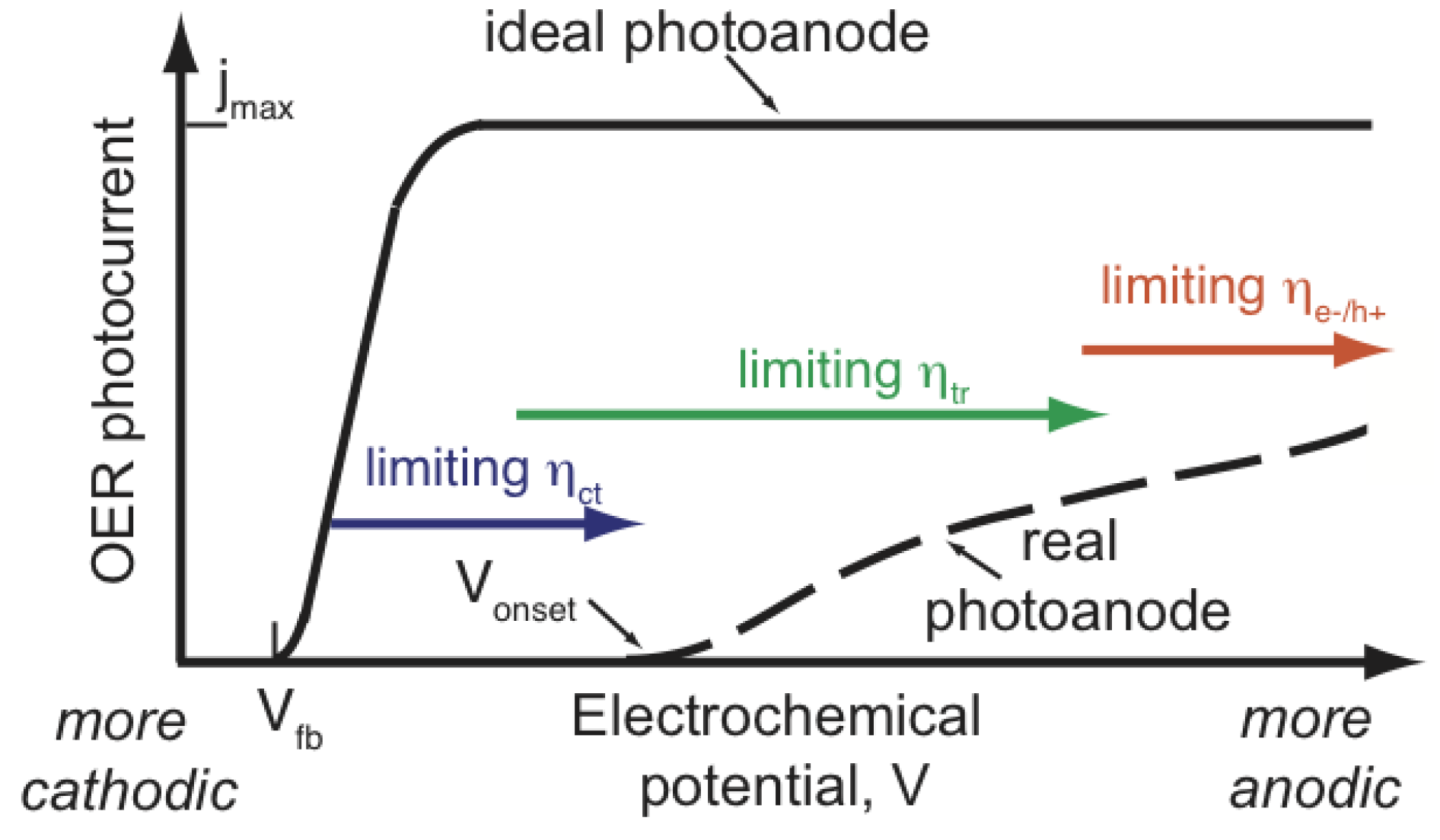
2.5. Overpotential
3. Conclusions
Acknowledgments
Conflicts of Interest
Appendix A. Theoretical Methodologies of Density Functional Theory [211,212]
Appendix A.1. The Hohenberg-Kohn Theorems
Appendix A.2. The Kohn-Sham Equations
Appendix A.3. The Exchange and Correlation Functionals
References
- Verne, J. L’Île Mystérieuse; Pierre-Jules Hetzel: Chartres, France, 1874. [Google Scholar]
- Campbell, C.J.; Laherrére, J.H. The End of Cheap Oil. Sci. Am. 1998, 278, 60–65. [Google Scholar] [CrossRef]
- Bentley, R.; Boyle, G. Global Oil Production: Forecasts and Methodologies. Environ. Plan. B Plan. Des. 2008, 35, 609–626. [Google Scholar] [CrossRef]
- Hook, M.; Tang, X. Depletion of fossil fuels and anthropogenic climate change—A review. Energy Policy 2013, 52, 797–809. [Google Scholar] [CrossRef]
- BP Statistical Review of World Energy. 2016. Available online: http://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html (accessed on 5 March 2017).
- Gerland, P.; Raftery, A.; Ševčíková, H.; Li, N.; Gu, D.; Spoorenberg, T.; Alkema, L.; Fosdick, B.; Chunn, J.; Lalic, N.; et al. World population stabilization unlikely this century. Science 2014, 346, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.A.; Warner, K.J. The 21st century population-energy-climate nexus. Energy Policy 2016, 93, 206–212. [Google Scholar] [CrossRef]
- Smalley, R.E. Future Global Energy Prosperity: The Terawatt Challenge. MRS Bull. 2005, 30, 412–417. [Google Scholar] [CrossRef]
- Lewis, N.S. Toward Cost-Effective Solar Energy Use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N.; Balzani, V. The Future of Energy Supply: Challenges and Opportunities. Angew. Chem. Int. Ed. 2007, 46, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Vesborg, P.C.K.; Jaramillo, T.F. Addressing the terawatt challenge: Scalability in the supply of chemical elements for renewable energy. RCS Adv. 2012, 2, 7933–7947. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed]
- Rajeshwar, K.; McConnell, R.; Harrison, K.; Licht, S. Solar Hydrogen Generation—Toward a Renewable Energy Future; Springer: Berlin, Germany, 2008. [Google Scholar]
- Herron, J.A.; Kim, J.; Upadhye, A.A.; Huber, G.W.; Maravelias, C.T. A general framework for the assessment of solar fuel technologies. Energy Environ. Sci. 2015, 8, 1754–5692. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I.; Naterer, G.F. Review of photocatalytic water-splitting methods for sustainable hydrogen production. Int. J. Energy Res. 2016, 40, 1449–1473. [Google Scholar] [CrossRef]
- Junge, H.; Rockstroh, N.; Fischer, S.; Brückner, A.; Ludwig, R.; Lochbrunner, S.; Kühn, O.; Beller, M. Light to Hydrogen: Photocatalytic Hydrogen Generation from Water with Molecularly-Defined Iron Complexes. Inorganics 2017, 5, 14. [Google Scholar] [CrossRef]
- Dura, L.; Wächtler, M.; Kupfer, S.; Kübel, J.; Ahrens, J.; Höfler, S.; Bröring, M.; Dietzek, B.; Beweries, T. Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction. Inorganics 2017, 5, 21. [Google Scholar] [CrossRef]
- Li, C.T.; Lin, R.Y.Y.; Lin, J.T. Sensitizers for Aqueous-Based Solar Cells. Chem. Asian J. 2017, 12, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.i.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef] [PubMed]
- Galliano, S.; Bella, F.; Gerbaldi, C.; Falco, M.; Viscardi, G.; Grätzel, M.; Barolo, C. Photoanode/Electrolyte Interface Stability in Aqueous Dye-Sensitized Solar Cells. Energy Technol. 2017, 5, 300–311. [Google Scholar] [CrossRef]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Gratzel, M.; Gerbaldi, C. Approaching truly sustainable solar cells by the use of water and cellulose derivatives. Green Chem. 2017, 19, 1043–1051. [Google Scholar] [CrossRef]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Gratzel, M.; Gerbaldi, C. Unveiling iodine-based electrolytes chemistry in aqueous dye-sensitized solar cells. Chem. Sci. 2016, 7, 4880–4890. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Pinaud, B.A.; Benck, J.D.; Seitz, L.C.; Forman, A.J.; Chen, Z.; Deutsch, T.G.; James, B.D.; Baum, K.N.; Baum, G.N.; Ardo, S.; et al. Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry. Energy Environ. Sci. 2013, 6, 1983–2002. [Google Scholar] [CrossRef]
- Jafari, T.; Moharreri, E.; Amin, A.S.; Miao, R.; Song, W.; Suib, S.L. Photocatalytic Water Splitting—The Untamed Dream: A Review of Recent Advances. Molecules 2016, 21, 900. [Google Scholar] [CrossRef] [PubMed]
- Navarro Yerga, R.; Álvarez Galván, M.; del Valle, F.; Villoria de la Mano, J.; Fierro, J. Water Splitting on Semiconductor Catalysts under Visible-Light Irradiation. ChemSusChem 2009, 2, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Fang, W.Q.; Zhao, H.J.; Yang, H.G. Inorganic Photocatalysts for Overall Water Splitting. Chem. Asian J. 2012, 7, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, F.E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 2013, 42, 2294–2320. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.; Zhang, J.; Yu, J.; Jaroniec, M.; Qiao, S.Z. Earth-abundant cocatalysts for semiconductor-based photocatalytic water splitting. Chem. Soc. Rev. 2014, 43, 7787–7812. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37. [Google Scholar] [CrossRef] [PubMed]
- Rajeshwar, K. Fundamentals of Semiconductor Electrochemistry and Photoelectrochemistry. In Encyclopedia of Electrochemistry; Wiley-VCH: Berlin, Germany, 2007. [Google Scholar]
- Van de Krol, R.; Grätzel, M. Photoelectrochemical Hydrogen Production; Springer: Berlin, Germany, 2012. [Google Scholar]
- Lewerenz, H.J.; Peter, L. Photoelectrochemical Water Splitting: Materials, Processes and Architectures; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Ni, M.; Leung, M.K.; Leung, D.Y.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Zhang, X.; Bieberle-Hütter, A. Modeling and Simulations in Photoelectrochemical Water Oxidation: From Single Level to Multiscale Modeling. ChemSusChem 2016, 9, 1223–1242. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 3. [Google Scholar] [CrossRef]
- Iandolo, B.; Wickman, B.; Zoric, I.; Hellman, A. The rise of hematite: Origin and strategies to reduce the high onset potential for the oxygen evolution reaction. J. Mater. Chem. A 2015, 3, 16896–16912. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Norskov, J.K. Materials for solar fuels and chemicals. Nat. Mater. 2017, 16, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Bak, T.; Nowotny, J.; Rekas, M.; Sorrell, C. Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects. Int. J. Hydrog. Energy 2002, 27, 991–1022. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef]
- Hasnip, P.J.; Refson, K.; Probert, M.I.J.; Yates, J.R.; Clark, S.J.; Pickard, C.J. Density functional theory in the solid state. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2014, 372, 20130270. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Dreizler, R.M.; Gross, E.K.U. Density-Functional Theory: An Approach to the Quantum Many-Body Problem; Springer: Berlin, Germany, 1990. [Google Scholar]
- Fiolhais, C.; Nogueira, F.; Marques, M. A Primer in Density Functional Theory; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Shin, Y.; Persson, K.A. Machine learning bandgaps of double perovskites. Nat. Rev. Mater. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Dai, H.L.; Ho, W. Spectroscopy and Photochemistry on Metal Surfaces; World Scientific: Singapore, Singapore, 1995. [Google Scholar]
- Liebsch, A. Electronic Excitations at Metal Surfaces; Plenum Press: Berlin, Germany, 1997. [Google Scholar]
- Nozik, A.J. Spectroscopy and hot electron relaxation dynamics in semiconductor quantum wells and quantum dots. Annu. Rev. Phys. Chem. 2001, 52, 193–231. [Google Scholar] [CrossRef] [PubMed]
- Brongersma, M.L.; Halas, N.J.; Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nano 2015, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.R.M.; Asenjo-Garcia, A.; García de Abajo, F.J. Hot-Electron Dynamics and Thermalization in Small Metallic Nanoparticles. ACS Photonics 2016, 3, 1637–1646. [Google Scholar] [CrossRef]
- Ogawa, T.; Yanai, N.; Monguzzi, A.; Kimizuka, N. Highly Efficient Photon Upconversion in Self-Assembled Light-Harvesting Molecular Systems. Sci. Rep. 2015, 5, 10882. [Google Scholar] [CrossRef] [PubMed]
- Godby, R.W.; Schlüter, M.; Sham, L.J. Trends in self-energy operators and their corresponding exchange-correlation potentials. Phys. Rev. B 1987, 36, 6497–6500. [Google Scholar] [CrossRef]
- Godby, R.W.; Schlüter, M.; Sham, L.J. Self-energy operators and exchange-correlation potentials in semiconductors. Phys. Rev. B 1988, 37, 10159–10175. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Kuisma, M.; Ojanen, J.; Enkovaara, J.; Rantala, T.T. Kohn-Sham potential with discontinuity for band gap materials. Phys. Rev. B 2010, 82, 115106. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796–A823. [Google Scholar] [CrossRef]
- Castelli, I.E.; Hüser, F.; Pandey, M.; Li, H.; Thygesen, K.S.; Seger, B.; Jain, A.; Persson, K.A.; Ceder, G.; Jacobsen, K.W. New Light-Harvesting Materials Using Accurate and Efficient Bandgap Calculations. Adv. Energy Mater. 2015, 5, 1400915. [Google Scholar] [CrossRef]
- Pilania, G.; Mannodi-Kanakkithodi, A.; Uberuaga, B.P.; Ramprasad, R.; Gubernatis, J.E.; Lookman, T. Machine learning bandgaps of double perovskites. Sci. Rep. 2016, 6, 19375. [Google Scholar] [CrossRef] [PubMed]
- Gallino, F.; Pacchioni, G.; Valentin, C.D. Transition levels of defect centers in ZnO by hybrid functionals and localized basis set approach. J. Chem. Phys. 2010, 133, 144512. [Google Scholar] [CrossRef] [PubMed]
- Lany, S.; Zunger, A. Assessment of correction methods for the band-gap problem and for finite-size effects in supercell defect calculations: Case studies for ZnO and GaAs. Phys. Rev. B 2008, 78, 235104. [Google Scholar] [CrossRef]
- De Walle, C.G.V.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into Current Limitations of Density Functional Theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014–4022. [Google Scholar] [CrossRef]
- Moss, T.S. The Interpretation of the Properties of Indium Antimonide. Proc. Phys. Soc. Sect. B 1954, 67, 775. [Google Scholar] [CrossRef]
- Burstein, E. Anomalous Optical Absorption Limit in InSb. Phys. Rev. 1954, 93, 632–633. [Google Scholar] [CrossRef]
- Vanpoucke, D.E.P.; Bultinck, P.; Cottenier, S.; Van Speybroeck, V.; Van Driessche, I. Aliovalent doping of CeO2: DFT study of oxidation state and vacancy effects. J. Mater. Chem. A 2014, 2, 13723–13737. [Google Scholar] [CrossRef]
- Celotti, G.; Nobili, D.; Ostoja, P. Lattice parameter study of silicon uniformly doped with boron and phosphorus. J. Mater. Sci. 1974, 9, 821–828. [Google Scholar] [CrossRef]
- Denton, A.R.; Ashcroft, N.W. Vegard’s law. Phys. Rev. A 1991, 43, 3161–3164. [Google Scholar] [CrossRef] [PubMed]
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Visible-Light Photocatalysis in Nitrogen-Doped Titanium Oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, S.; Paganini, M.C.; Giamello, E.; Selloni, A.; Di Valentin, C.; Pacchioni, G. Origin of Photoactivity of Nitrogen-Doped Titanium Dioxide under Visible Light. J. Am. Chem. Soc. 2006, 128, 15666–15671. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Reduced and n-Type Doped TiO2: Nature of Ti3+ Species. J. Phys. Chem. C 2009, 113, 20543–20552. [Google Scholar] [CrossRef]
- Kamisaka, H.; Adachi, T.; Yamashita, K. Theoretical study of the structure and optical properties of carbon-doped rutile and anatase titanium oxides. J. Chem. Phys. 2005, 123, 084704. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Theory of Carbon Doping of Titanium Dioxide. Chem. Mater. 2005, 17, 6656–6665. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, Y.; Sun, C.; Li, F.; Lu, G.; Cheng, H.M. Synergistic Effects of B/N Doping on the Visible-Light Photocatalytic Activity of Mesoporous TiO2. Angew. Chem. Int. Ed. 2008, 47, 4516–4520. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; English, N.J. First-Principles Calculation of Synergistic (N,P)-Codoping Effects on the Visible-Light Photocatalytic Activity of Anatase TiO2. J. Phys. Chem. C 2010, 114, 11984–11990. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Synergistic Effects of Bi/S Codoping on Visible Light-Activated Anatase TiO2 Photocatalysts from First Principles. J. Phys. Chem. C 2009, 113, 8373–8377. [Google Scholar] [CrossRef]
- Pelaez, M.; Nolan, N.T.; Pillai, S.C.; Seery, M.K.; Falaras, P.; Kontos, A.G.; Dunlop, P.S.; Hamilton, J.W.; Byrne, J.; O’Shea, K.; et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B Environ. 2012, 125, 331–349. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.Z.; Nagpure, S.; Kim, D.Y.; Rankin, S.E. Synthesis and Catalytic Applications of Non-Metal Doped Mesoporous Titania. Inorganics 2017, 5, 15. [Google Scholar] [CrossRef]
- Meng, X.Y.; Qin, G.W.; Li, S.; Wen, X.H.; Ren, Y.P.; Pei, W.L.; Zuo, L. Enhanced photoelectrochemical activity for Cu and Ti doped hematite: The first principles calculations. Appl. Phys. Lett. 2012, 98, 112104. [Google Scholar] [CrossRef]
- Liang, W.Y. Excitons. Phys. Educ. 1970, 5, 226. [Google Scholar] [CrossRef]
- Dember, H. Über eine photoelektronische Kraft in Kupferoxydul-Kristallen (Photoelectric E.M.F. in Cuprous-Oxide Crystals). Z. Phys. 1931, 32, 554. [Google Scholar]
- Sze, S.M. Semiconductor Devices, Physics and Technology; Wiley: Hoboken, NJ, USA, 1985. [Google Scholar]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Umezawa, H.; Ramanujam, K.; ichi Shikata, S. Thermally Stable Schottky Barrier Diode by Ru/Diamond. Appl. Phys. Express 2009, 2, 011202. [Google Scholar] [CrossRef]
- Kwon, S.; Lee, S.J.; Kim, S.M.; Lee, Y.; Song, H.; Park, J.Y. Probing the nanoscale Schottky barrier of metal/semiconductor interfaces of Pt/CdSe/Pt nanodumbbells by conductive-probe atomic force microscopy. Nanoscale 2015, 7, 12297–12301. [Google Scholar] [CrossRef] [PubMed]
- Durcan, C.A.; Balsano, R.; LaBella, V.P. Nanoscale mapping of the W/Si(001) Schottky barrier. J. Appl. Phys. 2014, 116, 023705. [Google Scholar] [CrossRef]
- Durcan, C.A.; Balsano, R.; LaBella, V.P. Time dependent changes in Schottky barrier mapping of the W/Si(001) interface utilizing ballistic electron emission microscopy. J. Appl. Phys. 2015, 117, 245306. [Google Scholar] [CrossRef]
- Tung, R.T. Electron transport at metal-semiconductor interfaces: General theory. Phys. Rev. B 1992, 45, 13509–13523. [Google Scholar] [CrossRef]
- Tung, R.T. Recent advances in Schottky barrier concepts. Mater. Sci. Eng. R Rep. 2001, 35, 1–138. [Google Scholar] [CrossRef]
- Tung, R.T. The physics and chemistry of the Schottky barrier height. Appl. Phys. Rev. 2014, 1, 011304. [Google Scholar]
- Jiao, Y.; Hellman, A.; Fang, Y.; Gao, S.; Käll, M. Schottky barrier formation and band bending revealed by first- principles calculations. Sci. Rep. 2015, 5, 11374. [Google Scholar] [CrossRef] [PubMed]
- Austin, I.; Mott, N. Polarons in crystalline and non-crystalline materials. Adv. Phys. 1969, 18, 41–102. [Google Scholar] [CrossRef]
- Shluger, A.L.; Stoneham, A.M. Small polarons in real crystals: Concepts and problems. J. Phys. Condens. Matter 1993, 5, 3049. [Google Scholar] [CrossRef]
- Bosman, A.; van Daal, H. Small-polaron versus band conduction in some transition-metal oxides. Adv. Phys. 1970, 19, 1–117. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation—Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Hellman, A.; Razaznejad, B.; Lundqvist, B.I. Potential-energy surfaces for excited states in extended systems. J. Chem. Phys. 2004, 120, 4593–4602. [Google Scholar] [CrossRef] [PubMed]
- Gavnholt, J.; Olsen, T.; Engelund, M.; Schiøtz, J. ΔSelf-consistent field method to obtain potential energy surfaces of excited molecules on surfaces. Phys. Rev. B 2008, 78, 075441. [Google Scholar] [CrossRef]
- Zawadzki, P.; Laursen, A.B.; Jacobsen, K.W.; Dahl, S.; Rossmeisl, J. Oxidative trends of TiO2-hole trapping at anatase and rutile surfaces. Energy Environ. Sci. 2012, 5, 9866–9869. [Google Scholar] [CrossRef]
- Lany, S.; Zunger, A. Polaronic hole localization and multiple hole binding of acceptors in oxide wide-gap semiconductors. Phys. Rev. B 2009, 80, 085202. [Google Scholar] [CrossRef]
- Alexandrov, V.; Neumann, A.; Scherer, M.M.; Rosso, K.M. Electron Exchange and Conduction in Nontronite from First-Principles. J. Phys. Chem. C 2013, 117, 2032–2040. [Google Scholar] [CrossRef]
- Maxisch, T.; Zhou, F.; Ceder, G. Ab initio study of the migration of small polarons in olivine LixFePO4 and their association with lithium ions and vacancies. Phys. Rev. B 2006, 73, 104301. [Google Scholar] [CrossRef]
- Chen, H.; Umezawa, N. Hole localization, migration, and the formation of peroxide anion in perovskite SrTiO3. Phys. Rev. B 2014, 90, 035202. [Google Scholar] [CrossRef]
- Carvalho, A.; Alkauskas, A.; Pasquarello, A.; Tagantsev, A.K.; Setter, N. A hybrid density functional study of lithium in ZnO: Stability, ionization levels, and diffusion. Phys. Rev. B 2009, 80, 195205. [Google Scholar] [CrossRef]
- Zawadzki, P.; Jacobsen, K.W.; Rossmeisl, J. Electronic hole localization in rutile and anatase TiO2- Self-interaction correction in Δ-SCF {DFT}. Chem. Phys. Lett. 2011, 506, 42–45. [Google Scholar] [CrossRef]
- Deskins, N.A.; Rousseau, R.; Dupuis, M. Localized Electronic States from Surface Hydroxyls and Polarons in TiO2(110). J. Phys. Chem. C 2009, 113, 14583–14586. [Google Scholar] [CrossRef]
- Deskins, N.A.; Dupuis, M. Intrinsic Hole Migration Rates in TiO2 from Density Functional Theory. J. Phys. Chem. C 2009, 113, 346–358. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Holstein, T. Studies of Polaron Motion: Part I. The Molecular-Crystal Model. Ann. Phys. 2000, 8, 706–724. [Google Scholar] [CrossRef]
- Holstein, T. Studies of Polaron Motion: Part II. The “Small” Polaron. Ann. Phys. 2000, 281, 725–773. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Electronic Structure of Defect States in Hydroxylated and Reduced Rutile TiO2(110) Surfaces. Phys. Rev. Lett. 2006, 97, 166803. [Google Scholar] [CrossRef] [PubMed]
- Hurum, D.C.; Agrios, A.G.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Explaining the Enhanced Photocatalytic Activity of Degussa P25 Mixed-Phase TiO2 Using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, B.; Luo, Y. Location of Trapped Hole on Rutile-TiO2(110) Surface and Its Role in Water Oxidation. J. Phys. Chem. C 2012, 116, 7863–7866. [Google Scholar] [CrossRef]
- Kleiman-Shwarsctein, A.; Huda, M.N.; Walsh, A.; Yan, Y.; Stucky, G.D.; Hu, Y.S.; Al-Jassim, M.M.; McFarland, E.W. Electrodeposited Aluminum-Doped α-Fe2O3 Photoelectrodes: Experiment and Theory. Chem. Mater. 2010, 22, 510–517. [Google Scholar] [CrossRef]
- Liao, P.; Carter, E.A. Hole transport in pure and doped hematite. J. Appl. Phys. 2012, 112, 013701. [Google Scholar] [CrossRef]
- Adelstein, N.; Neaton, J.B.; Asta, M.; De Jonghe, L.C. Density functional theory based calculation of small-polaron mobility in hematite. Phys. Rev. B 2014, 89, 245115. [Google Scholar] [CrossRef]
- Pacchioni, G. Modeling doped and defective oxides in catalysis with density functional theory methods: Room for improvements. J. Chem. Phys. 2008, 128, 182505. [Google Scholar] [CrossRef] [PubMed]
- Shluger, A.L.; McKenna, K.P.; Sushko, P.V.; Ramo, D.M.; Kimmel, A.V. Modelling of electron and hole trapping in oxides. Model. Simul. Mater. Sci. Eng. 2009, 17, 084004. [Google Scholar] [CrossRef]
- Zou, Z.; Ye, J.; Sayama, K.; Arakawa, H. Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst. Nature 2001, 414, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.; Abdullah, H. Electron transport study of indium oxide as photoanode in DSSCs: A review. Renew. Sustain. Energy Rev. 2016, 63, 245–255. [Google Scholar] [CrossRef]
- Frank, A.J.; Kopidakis, N.; van de Lagemaat, J. Electrons in nanostructured TiO2 solar cells: Transport, recombination and photovoltaic properties. Coord. Chem. Rev. 2004, 248, 1165–1179. [Google Scholar] [CrossRef]
- Riss, A.; Elser, M.J.; Bernardi, J.; Diwald, O. Stability and Photoelectronic Properties of Layered Titanate Nanostructures. J. Am. Chem. Soc. 2009, 131, 6198–6206. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, P.; Sun, Y.; Wu, Y.; Mayers, B.; Gates, B.; Yin, Y.; Kim, F.; Yan, H. One-Dimensional Nanostructures: Synthesis, Characterization, and Applications. Adv. Mater. 2003, 15, 353–389. [Google Scholar] [CrossRef]
- Anta, J.A.; Nelson, J.; Quirke, N. Charge transport model for disordered materials: Application to sensitized TiO2. Phys. Rev. B 2002, 65, 125324. [Google Scholar] [CrossRef]
- Göpel, W.; Rocker, G.; Feierabend, R. Intrinsic defects of TiO2(110): Interaction with chemisorbed O2, H2, CO, and CO2. Phys. Rev. B 1983, 28, 3427–3438. [Google Scholar] [CrossRef]
- Wickman, B.; Bastos Fanta, A.; Burrows, A.; Hellman, A.; Wagner, J.B.; Iandolo, B. Iron Oxide Films Prepared by Rapid Thermal Processing for Solar Energy Conversion. Sci. Rep. 2017, 7, 40500. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Z.; Li, W.; Zhang, X.; He, D.; Xiao, X. Ag Nanoparticles Located on Three-Dimensional Pine Tree-Like Hierarchical TiO2 Nanotube Array Films as High-Efficiency Plasmonic Photocatalysts. Nanoscale Res. Lett. 2017, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Antony, A.C.; Akhade, S.A.; Liang, T.; Janik, M.J.; Maranas, J.K.; Sinnott, S.B. Simulating an Applied Voltage in Molecular Dynamics Using Charge Optimized Many Body (COMB3) Potentials. ECS Trans. 2015, 69, 103–105. [Google Scholar] [CrossRef]
- Van de Lagemaat, J.; Park, N.G.; Frank, A.J. Influence of Electrical Potential Distribution, Charge Transport, and Recombination on the Photopotential and Photocurrent Conversion Efficiency of Dye-Sensitized Nanocrystalline TiO2 Solar Cells: A Study by Electrical Impedance and Optical Modulation Techniques. J. Phys. Chem. B 2000, 104, 2044–2052. [Google Scholar] [CrossRef]
- De Jongh, P.E.; Vanmaekelbergh, D. Trap-Limited Electronic Transport in Assemblies of Nanometer-Size TiO2 Particles. Phys. Rev. Lett. 1996, 77, 3427–3430. [Google Scholar] [CrossRef] [PubMed]
- Vanmaekelbergh, D.; de Jongh, P.E. Electron transport in disordered semiconductors studied by a small harmonic modulation of the steady state. Phys. Rev. B 2000, 61, 4699–4704. [Google Scholar] [CrossRef]
- Anderson, A.B.; Albu, T.V. Ab Initio Determination of Reversible Potentials and Activation Energies for Outer-Sphere Oxygen Reduction to Water and the Reverse Oxidation Reaction. J. Am. Chem. Soc. 1999, 121, 11855–11863. [Google Scholar] [CrossRef]
- Anderson, A.B.; Neshev, N.M.; Sidik, R.A.; Shiller, P. Mechanism for the electrooxidation of water to {OH} and O bonded to platinum: Quantum chemical theory. Electrochim. Acta 2002, 47, 2999–3008. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Anderson, A.B. Aqueous and Surface Redox Potentials from Self-Consistently Determined Gibbs Energies. J. Phys. Chem. C 2008, 112, 8747–8750. [Google Scholar] [CrossRef]
- Anderson, A.B. Insights into electrocatalysis. Phys. Chem. Chem. Phys. 2012, 14, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.B. Theory at the electrochemical interface: Reversible potentials and potential-dependent activation energies. Electrochim. Acta 2003, 48, 3743–3749. [Google Scholar] [CrossRef]
- Anderson, A.B. Theories for Predicting Reversible Potentials of Reactions on Electrode Surfaces from Internal and Gibbs Energies: Applications to ORR. ECS Trans. 2010, 28, 1–17. [Google Scholar]
- Taylor, C.D.; Kelly, R.G.; Neurock, M. A first-principles analysis of the chemisorption of hydroxide on copper under electrochemical conditions: A probe of the electronic interactions that control chemisorption at the electrochemical interface. J. Electroanal. Chem. 2007, 607, 167–174. [Google Scholar] [CrossRef]
- Taylor, C.; Kelly, R.G.; Neurock, M. First-Principles Calculations of the Electrochemical Reactions of Water at an Immersed Ni (111)/H2O Interface. J. Electrochem. Soc. 2006, 153, E207–E214. [Google Scholar] [CrossRef]
- Taylor, C.D.; Kelly, R.G.; Neurock, M. First-Principles Prediction of Equilibrium Potentials for Water Activation by a Series of Metals. J. Electrochem. Soc. 2007, 154, F217–F221. [Google Scholar] [CrossRef]
- Janik, M.J.; Taylor, C.D.; Neurock, M. First-Principles Analysis of the Initial Electroreduction Steps of Oxygen over Pt(111). J. Electrochem. Soc. 2009, 156, B126–B135. [Google Scholar] [CrossRef]
- Taylor, C.; Kelly, R.G.; Neurock, M. Theoretical Analysis of the Nature of Hydrogen at the Electrochemical Interface Between Water and a Ni(111) Single-Crystal Electrode. J. Electrochem. Soc. 2007, 154, F55–F64. [Google Scholar] [CrossRef]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Bondarenko, A.S.; Stephens, I.E.L.; Hansen, H.A.; Pérez-Alonso, F.J.; Tripkovic, V.; Johansson, T.P.; Rossmeisl, J.; Nørskov, J.K.; Chorkendorff, I. The Pt(111)/Electrolyte Interface under Oxygen Reduction Reaction Conditions: An Electrochemical Impedance Spectroscopy Study. Langmuir 2011, 27, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated Phase Diagrams for the Electrochemical Oxidation and Reduction of Water over Pt(111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Rossmeisl, J.; Hellman, A.; Nørskov, J.K. Theoretical Trends in Particle Size Effects for the Oxygen Reduction Reaction. Z. Phys. Chem. 2007, 221, 1209–1220. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Nielsen, M.; Björketun, M.E.; Hansen, M.H.; Rossmeisl, J. Towards first principles modeling of electrochemical electrode–electrolyte interfaces. Surf. Sci. 2015, 613, 2–7. [Google Scholar] [CrossRef]
- Valdés, Á.; Qu, Z.-W.; Kroes, G.-J.; Rossmeisl, J.; Nørskov, J.K. Oxidation and Photo-Oxidation of Water on TiO2 Surface. J. Phys. Chem. C 2008, 112, 9872–9879. [Google Scholar] [CrossRef]
- Valdés, Á.; Kroes, G.J. First principles study of the photo-oxidation of water on tungsten trioxide (WO3). J. Chem. Phys. 2009, 130, 114701. [Google Scholar] [CrossRef] [PubMed]
- Valdés, Á.; Kroes, G.J. Cluster Study of the Photo-Oxidation of Water on Rutile Titanium Dioxide (TiO2). J. Phys. Chem. C 2010, 114, 1701–1708. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T. First-principles computational electrochemistry: Achievements and challenges. Electrochim. Acta 2012, 84, 3–11. [Google Scholar] [CrossRef]
- Karlberg, G.S.; Rossmeisl, J.; Norskov, J.K. Estimations of electric field effects on the oxygen reduction reaction based on the density functional theory. Phys. Chem. Chem. Phys. 2007, 9, 5158–5161. [Google Scholar] [CrossRef] [PubMed]
- Fenter, P.; Sturchio, N.C. Mineral-water interfacial structures revealed by synchrotron X-ray scattering. Prog. Surf. Sci. 2004, 77, 171–258. [Google Scholar] [CrossRef]
- Sato, N. Electrochemistry at Metal and Semiconductor Electrodes; Elsevier: Oxford, UK, 1998. [Google Scholar]
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Simulating an Applied Voltage in Molecular Dynamics Using Charge Optimized Many Body (COMB3) Potentials. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar] [CrossRef] [PubMed]
- Skulason, E.; Karlberg, G.S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jonsson, H.; Norskov, J.K. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Phys. Chem. Chem. Phys. 2007, 9, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.Y.; Wasileski, S.A.; Janik, M.J. Electronic structure models of oxygen adsorption at the solvated, electrified Pt(111) interface. Phys. Chem. Chem. Phys. 2009, 11, 10108–10117. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.Y.; Janik, M.J.; Maranas, J.K. Molecular dynamics simulations of an electrified water/Pt(111) interface using point charge dissociative water. Electrochim. Acta 2013, 101, 308–325. [Google Scholar] [CrossRef]
- Hansen, M.H.; Jin, C.; Thygesen, K.S.; Rossmeisl, J. Finite Bias Calculations to Model Interface Dipoles in Electrochemical Cells at the Atomic Scale. J. Phys. Chem. C 2016, 120, 13485–13491. [Google Scholar] [CrossRef]
- Zhang, C.; Sprik, M. Finite field methods for the supercell modeling of charged insulator/electrolyte interfaces. Phys. Rev. B 2016, 94, 245309. [Google Scholar] [CrossRef]
- Russo, D.; Teixeira, J.; Kneller, L.; Copley, J.R.D.; Ollivier, J.; Perticaroli, S.; Pellegrini, E.; Gonzalez, M.A. Vibrational Density of States of Hydration Water at Biomolecular Sites: Hydrophobicity Promotes Low Density Amorphous Ice Behavior. J. Am. Chem. Soc. 2011, 133, 4882–4888. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Neogi, S.; Kent, P.R.C.; Bandura, A.V.; Kubicki, J.D.; Wesolowski, D.J.; Cole, D.; Sofo, J.O. Hydrogen Bonds and Vibrations of Water on (110) Rutile. J. Phys. Chem. C 2009, 113, 13732–13740. [Google Scholar] [CrossRef]
- English, N.J.; Kavathekar, R.S.; MacElroy, J. Hydrogen bond dynamical properties of adsorbed liquid water monolayers with various TiO2 interfaces. Mol. Phys. 2012, 110, 2919–2925. [Google Scholar] [CrossRef]
- Kavathekar, R.S.; Dev, P.; English, N.J.; MacElroy, J. Molecular dynamics study of water in contact with the TiO2 rutile-110, 100, 101, 001 and anatase-101, 001 surface. Mol. Phys. 2011, 109, 1649–1656. [Google Scholar] [CrossRef]
- Iandolo, B.; Hellman, A. The Role of Surface States in the Oxygen Evolution Reaction on Hematite. Angew. Chem. Int. Ed. 2014, 53, 13404–13408. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Pala, R.G.S. First-Principles Study of Photoinduced Water-Splitting on Fe2O3. J. Phys. Chem. C 2011, 115, 12901–12907. [Google Scholar] [CrossRef]
- Hellman, A.; Iandolo, B.; Wickman, B.; Grönbeck, H.; Baltrusaitis, J. Electro-oxidation of water on hematite: Effects of surface termination and oxygen vacancies investigated by first-principles. Surf. Sci. 2015, 640, 45–49. [Google Scholar] [CrossRef]
- Hansen, H.A.; Rossmeisl, J.; Norskov, J.K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni(111) surfaces studied by DFT). Phys. Chem. Chem. Phys. 2008, 10, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Persson, K.A.; Waldwick, B.; Lazic, P.; Ceder, G. Prediction of solid-aqueous equilibria: Scheme to combine first-principles calculations of solids with experimental aqueous states. Phys. Rev. B 2012, 85, 235438. [Google Scholar] [CrossRef]
- Todorova, M.; Neugebauer, J. Extending the Concept of Defect Chemistry from Semiconductor Physics to Electrochemistry. Phys. Rev. Appl. 2014, 1, 014001. [Google Scholar] [CrossRef]
- Huang, L.F.; Rondinelli, J.M. Electrochemical phase diagrams for Ti oxides from density functional calculations. Phys. Rev. B 2015, 92, 245126. [Google Scholar] [CrossRef]
- Zeng, Z.; Chan, M.K.Y.; Zhao, Z.J.; Kubal, J.; Fan, D.; Greeley, J. Towards First Principles-Based Prediction of Highly Accurate Electrochemical Pourbaix Diagrams. J. Phys. Chem. C 2015, 119, 18177–18187. [Google Scholar] [CrossRef]
- Ulissi, Z.W.; Singh, A.R.; Tsai, C.; Nørskov, J.K.; Todorova, M.; Neugebauer, J. Automated Discovery and Construction of Surface Phase Diagrams Using Machine Learning. J. Phys. Chem. Lett. 2016, 7, 3931–3935. [Google Scholar] [CrossRef] [PubMed]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; National Association of Corrosion Engineers: Houston, TX, USA, 1974. [Google Scholar]
- Nguyen, M.T.; Piccinin, S.; Seriani, N.; Gebauer, R. Photo-Oxidation of Water on Defective Hematite(0001). ACS Catal. 2015, 5, 715–721. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Bahn, S.; Hansen, L.B.; Bollinger, M.; Bengaard, H.; Hammer, B.; Sljivancanin, Z.; Mavrikakis, M.; et al. Universality in Heterogeneous Catalysis. J. Catal. 2002, 209, 275. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Seriani, N.; Gebauer, R. Water adsorption and dissociation on α-Fe2O3(0001): PBE+U calculations. J. Chem. Phys. 2013, 138, 194709. [Google Scholar] [CrossRef] [PubMed]
- Toroker, M.C. Theoretical Insights into the Mechanism of Water Oxidation on Nonstoichiometric and Titanium-Doped Fe2O3(0001). J. Phys. Chem. C 2014, 118, 23162–23167. [Google Scholar] [CrossRef]
- Zeng, Z.; Hansen, M.H.; Greeley, J.P.; Rossmeisl, J.; Björketun, M.E. Ab Initio Thermodynamic Modeling of Electrified Metal–Oxide Interfaces: Consistent Treatment of Electronic and Ionic Chemical Potentials. J. Phys. Chem. C 2014, 118, 22663–22671. [Google Scholar] [CrossRef]
- Chan, K.; Nørskov, J.K. Electrochemical Barriers Made Simple. J. Phys. Chem. Lett. 2015, 6, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skúlason, E.; Bligaard, T.; Nørskov, J.K. Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces. Phys. Rev. Lett. 2007, 99, 016105. [Google Scholar] [CrossRef] [PubMed]
- Bligaard, T.; Nørskov, J.; Dahl, S.; Matthiesen, J.; Christensen, C.; Sehested, J. The Brønsted–Evans–Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Norskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- García-Mota, M.; Vojvodic, A.; Metiu, H.; Man, I.C.; Su, H.Y.; Rossmeisl, J.; Nørskov, J.K. Tailoring the Activity for Oxygen Evolution Electrocatalysis on Rutile TiO2(110) by Transition-Metal Substitution. ChemCatChem 2011, 3, 1607–1611. [Google Scholar] [CrossRef]
- Montoya, J.H.; Garcia-Mota, M.; Norskov, J.K.; Vojvodic, A. Theoretical evaluation of the surface electrochemistry of perovskites with promising photon absorption properties for solar water splitting. Phys. Chem. Chem. Phys. 2015, 17, 2634–2640. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Martinez, J.I.; Rossmeisl, J. Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions. Phys. Chem. Chem. Phys. 2011, 13, 15639–15643. [Google Scholar] [CrossRef] [PubMed]
- Dogutan, D.K.; Bediako, D.K.; Teets, T.S.; Schwalbe, M.; Nocera, D.G. Efficient Synthesis of Hangman Porphyrins. Org. Lett. 2010, 12, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- McGuire R., Jr.; Dogutan, D.K.; Teets, T.S.; Suntivich, J.; Shao-Horn, Y.; Nocera, D.G. Oxygen reduction reactivity of cobalt(II) hangman porphyrins. Chem. Sci. 2010, 1, 411–414. [Google Scholar] [CrossRef]
- Rosenthal, J.; Nocera, D. Oxygen activation chemistry of Pacman and Hangman porphyrin architectures based on xanthene and dibenzofuran spacers. In Progress in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 2007; Volume 55, pp. 483–544. [Google Scholar]
- Dogutan, D.K.; Stoian, S.A.; McGuire, R.; Schwalbe, M.; Teets, T.S.; Nocera, D.G. Hangman Corroles: Efficient Synthesis and Oxygen Reaction Chemistry. J. Am. Chem. Soc. 2011, 133, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chng, L.L.; Nocera, D.G. Proton-Coupled O–O Activation on a Redox Platform Bearing a Hydrogen-Bonding Scaffold. J. Am. Chem. Soc. 2003, 125, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Baran, J.D.; Grönbeck, H.; Hellman, A. Analysis of Porphyrines as Catalysts for Electrochemical Reduction of O2 and Oxidation of H2O. J. Am. Chem. Soc. 2014, 136, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Que, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Shook, R.L.; Borovik, A.S. Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem. 2010, 49, 3646–3660. [Google Scholar] [CrossRef] [PubMed]
- Wang, B. Electronic Structure and Optical Properties of Solar Energy Materials. Ph.D. Dissertation, Royal Institute of Technology, KTH, Stockholm, Sweden, 2014. [Google Scholar]
- Martin, R.M. Electronic Structure : Basic Theory and Practical Methods; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Slater, J.C. A Simplification of the Hartree-Fock Method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 2009, 79, 155107. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Tests of a ladder of density functionals for bulk solids and surfaces. Phys. Rev. B 2004, 69, 075102. [Google Scholar] [CrossRef]
- Perdew, J.P. Accurate Density Functional for the Energy: Real-Space Cutoff of the Gradient Expansion for the Exchange Hole. Phys. Rev. Lett. 1985, 55, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Langreth, D.C.; Mehl, M.J. Easily Implementable Nonlocal Exchange-Correlation Energy Functional. Phys. Rev. Lett. 1981, 47, 446–450. [Google Scholar] [CrossRef]
- Gunnarson, O.; Lundqvist, B.; Lundqvist, S. Screening in a spin-polarized electron liquid. Solid State Commun. 1972, 11, 149–153. [Google Scholar] [CrossRef]
- Beck, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Armiento, R.; Mattsson, A.E. Functional designed to include surface effects in self-consistent density functional theory. Phys. Rev. B 2005, 72, 085108. [Google Scholar] [CrossRef]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta? Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Constantin, L.A.; Sun, J. Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry. Phys. Rev. Lett. 2009, 103, 026403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree Fock and local density functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hellman, A.; Wang, B. First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study. Inorganics 2017, 5, 37. https://doi.org/10.3390/inorganics5020037
Hellman A, Wang B. First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study. Inorganics. 2017; 5(2):37. https://doi.org/10.3390/inorganics5020037
Chicago/Turabian StyleHellman, Anders, and Baochang Wang. 2017. "First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study" Inorganics 5, no. 2: 37. https://doi.org/10.3390/inorganics5020037
APA StyleHellman, A., & Wang, B. (2017). First-Principles View on Photoelectrochemistry: Water-Splitting as Case Study. Inorganics, 5(2), 37. https://doi.org/10.3390/inorganics5020037