
Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst



Abstract
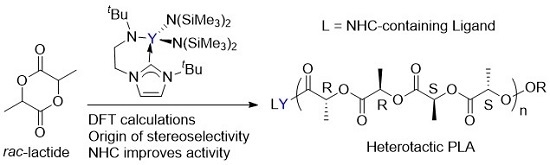
:
1. Introduction
2. Computational Details
3. Results and Discussion
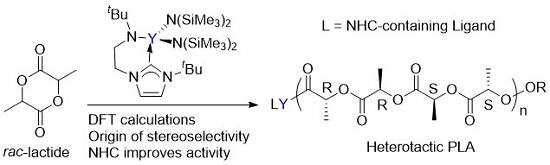
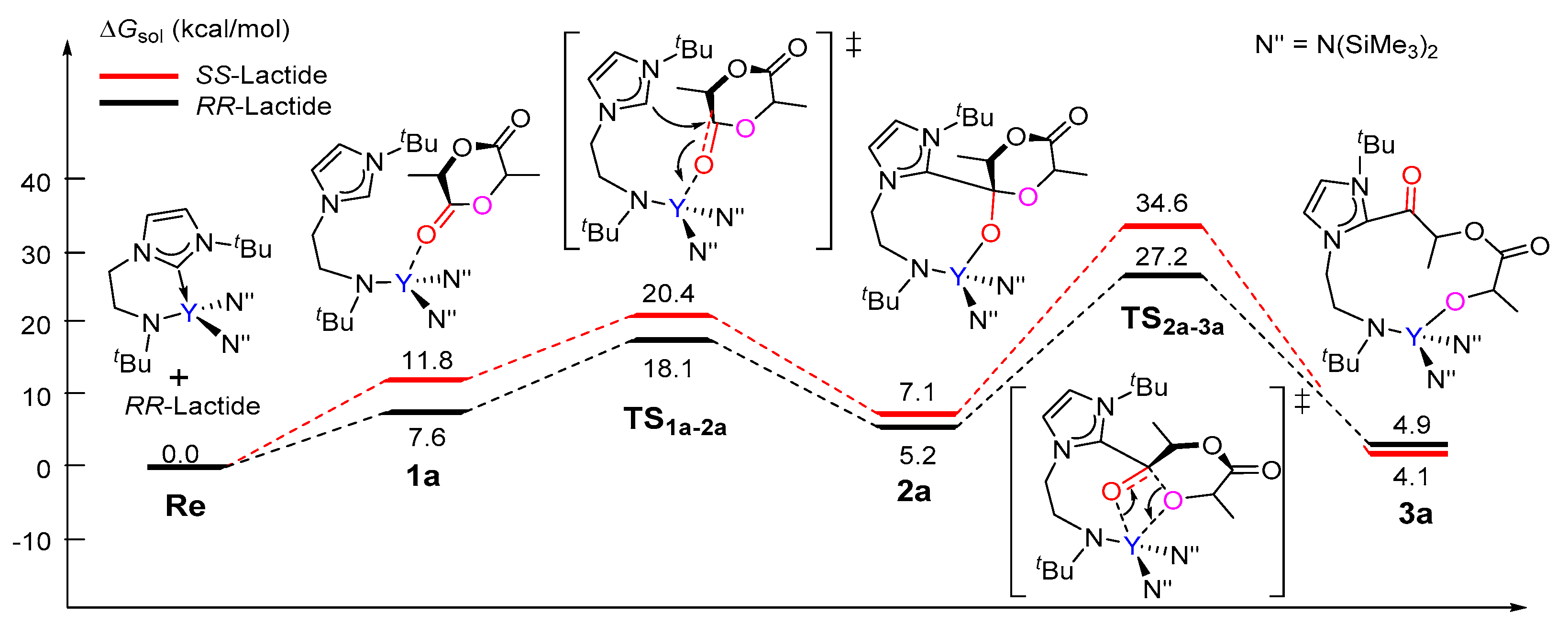
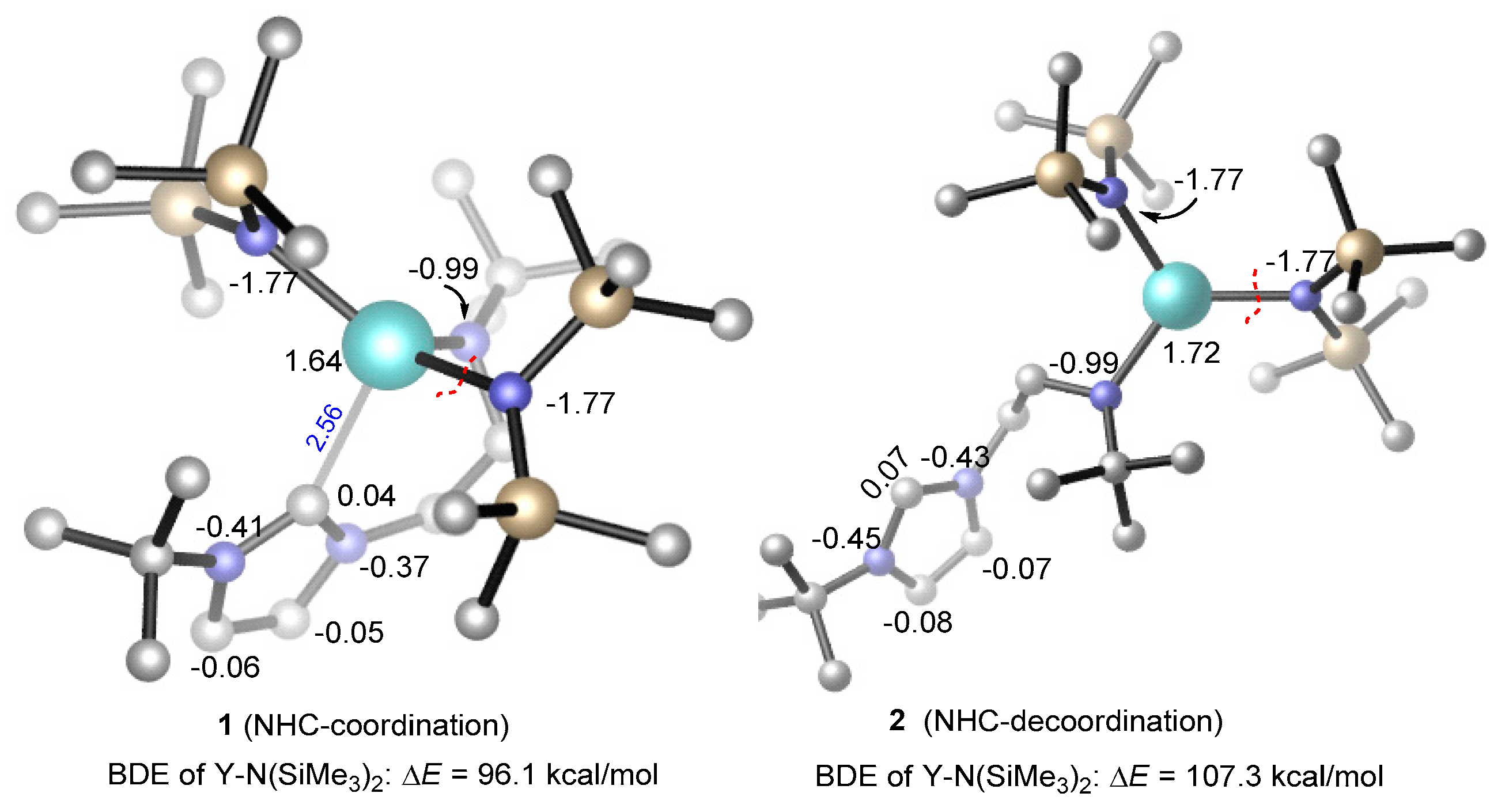
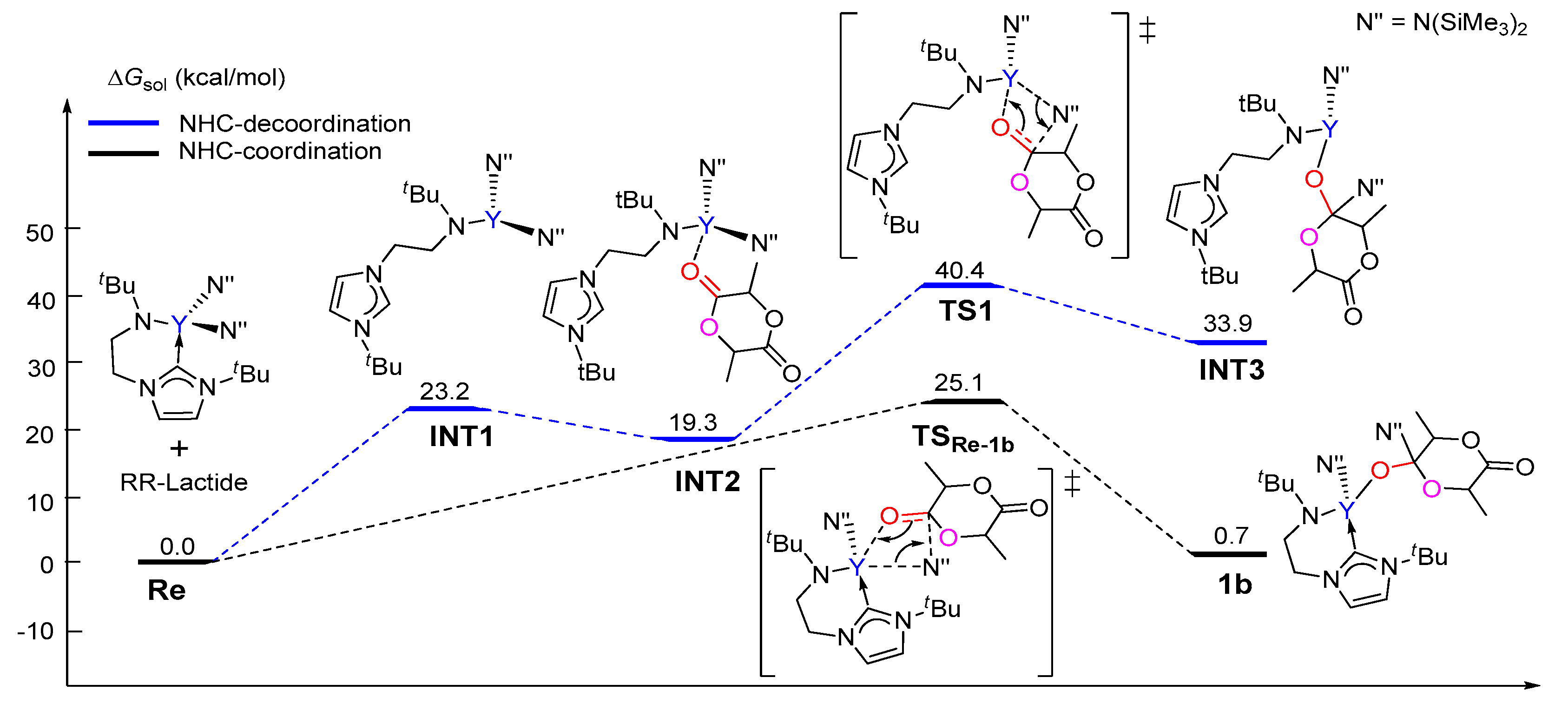
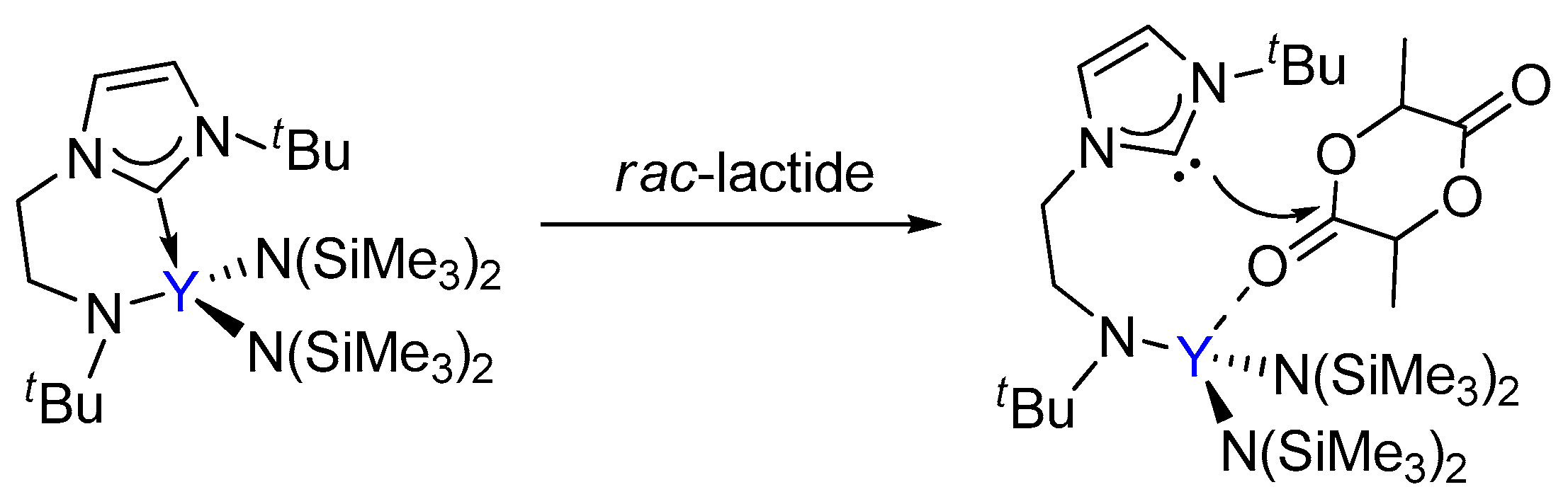
3.1. The Chain Initiation Step of Lactide ROP Mediated by the Bifunctional Yttrium Complex
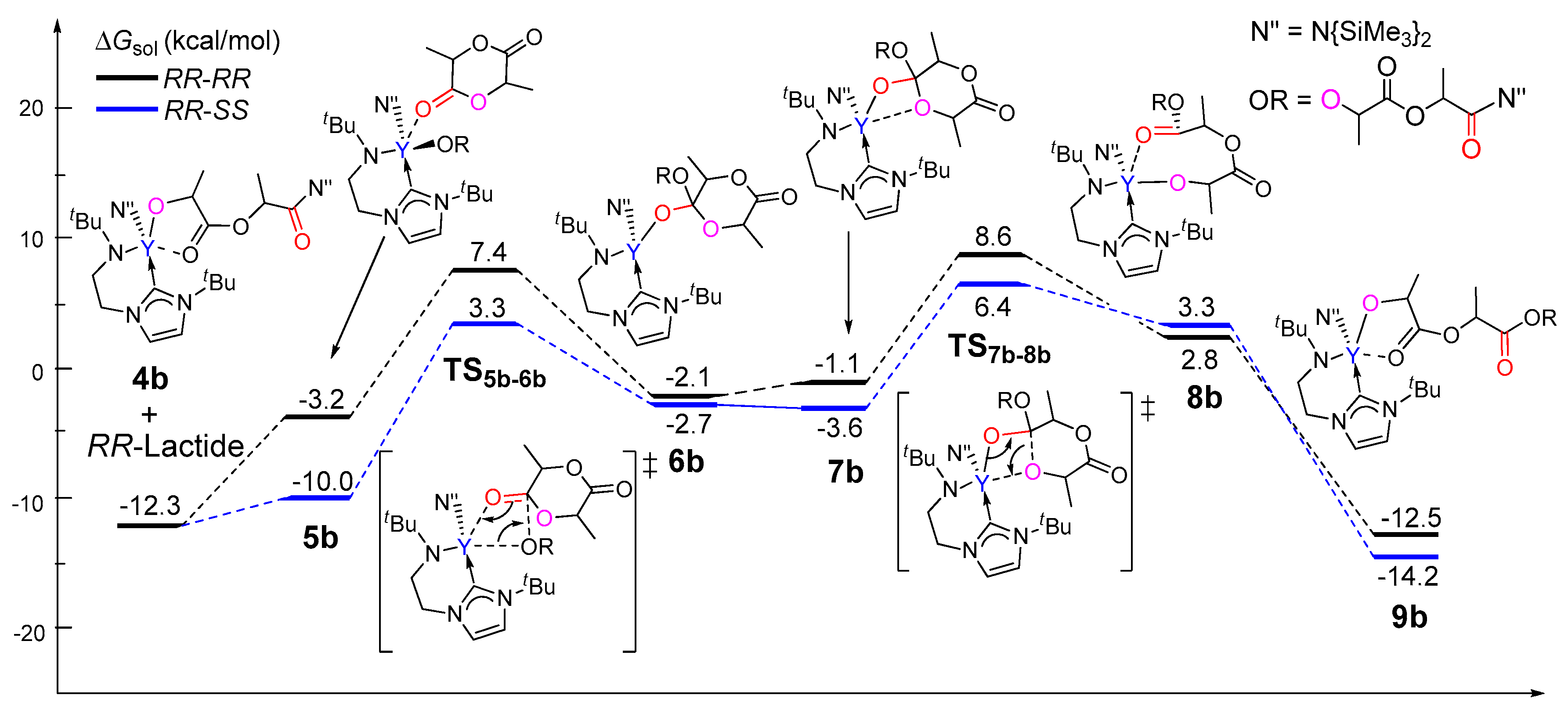
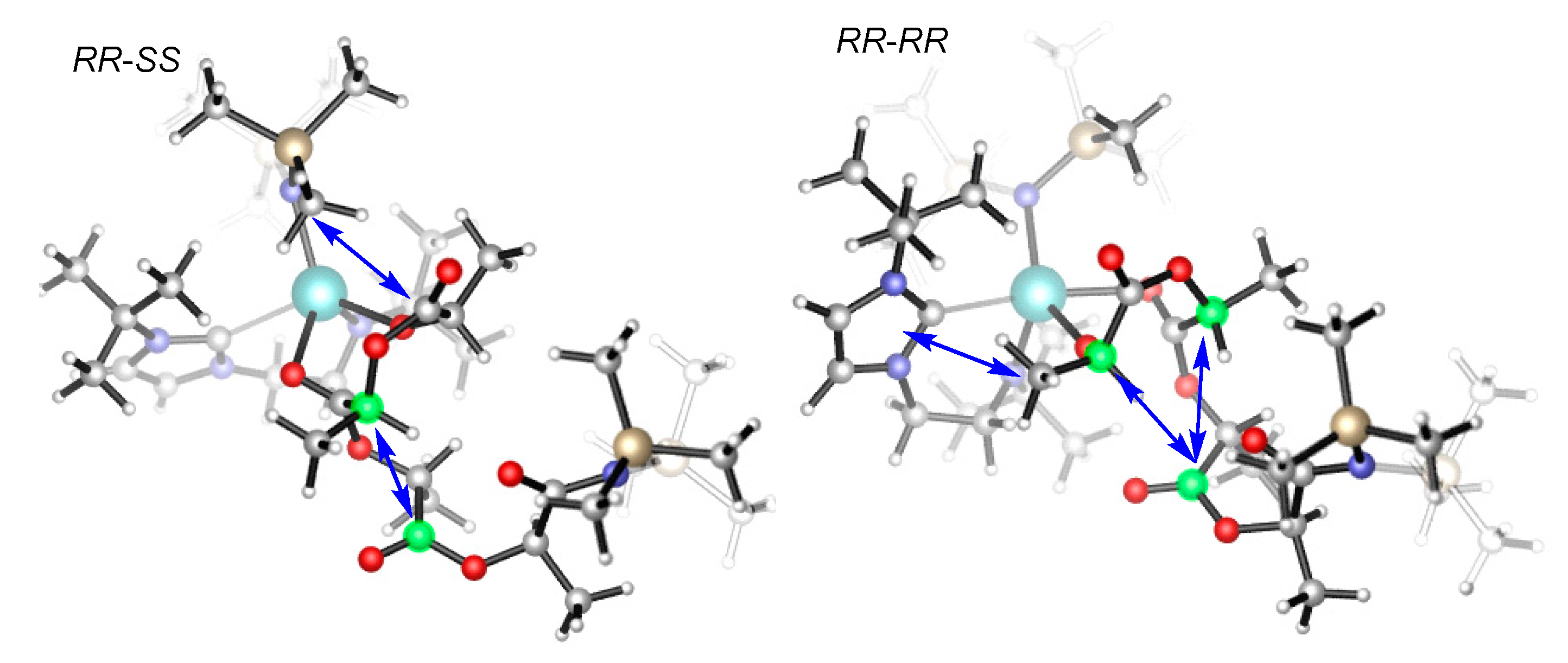
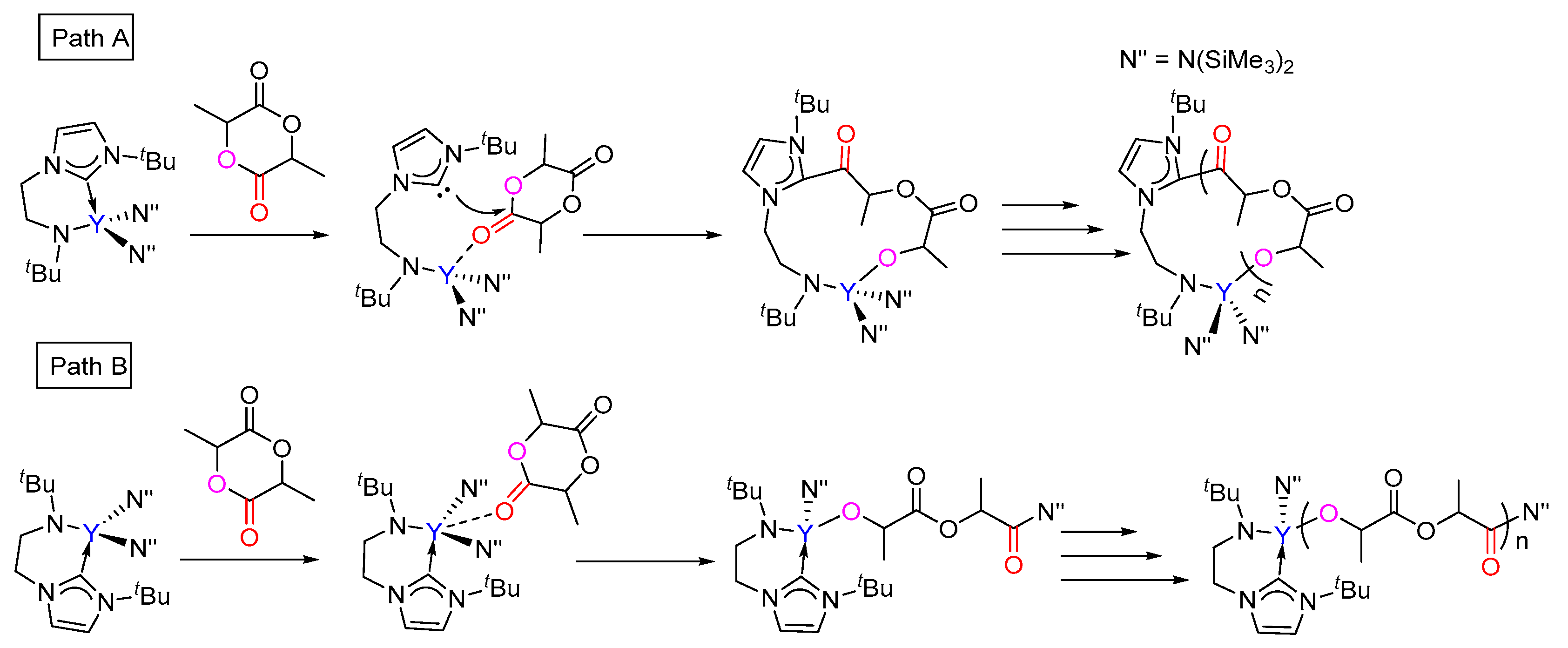
3.2. Selectivity of rac-Lactide Polymerization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.P.; Kumar, V. New emerging trends in synthetic biodegradable polymers—Polylactide: A critique. Eur. Polym. J. 2007, 43, 4053–4074. [Google Scholar] [CrossRef]
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Liu, X.; Shang, X. Achiral lanthanide alkyl complexes bearing N,O multidentate ligands. Synthesis and catalysis of highly heteroselective ring-opening polymerization of rac-lactide. Organometallics 2007, 26, 2747–2757. [Google Scholar]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J., Jr.; Hallett, J.P.; Leak, D.J.; Liotta, C.L. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Platel, R.H.; Hodgson, L.M.; Williams, C.K. Biocompatible initiators for lactide polymerization. Polym. Rev. 2008, 48, 11–63. [Google Scholar] [CrossRef]
- Stanford, M.J.; Dove, A.P. Stereocontrolled ring-opening polymerisation of lactide. Chem. Soc. Rev. 2010, 39, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Rosen, T.; Goldberg, I.; Venditto, K.M. Tailor-made stereoblock copolymers of poly(lactic acid) by a truly living polymerization catalyst. J. Am. Chem. Soc. 2016, 138, 12041–12044. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schmid, T.E.; Richard, V.; Haquette, P.; Raman, S.K.; Rager, M.N.; Gauvin, R.M.; Morin, Y.; Trivelli, X.; Guerineau, V. Mechanistic aspects of the polymerization of lactide using a highly efficient aluminum(III) catalytic system. J. Am. Chem. Soc. 2017, 139, 6217–6225. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Crisci, A.G.D.; Hedrick, J.L.; Waymouth, R.M. Amidine-mediated zwitterionic polymerization of lactide. ACS Macro Lett. 2012, 1, 1113–1115. [Google Scholar] [CrossRef]
- Dove, A.P. Organic Catalysis for Ring-Opening Polymerization. ACS Macro Lett. 2012, 1, 1409–1412. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Dove, A.P. Controlled ring-opening polymerisation of cyclic esters: Polymer blocks in self. Chem. Commun. 2008, 48, 6446–6470. [Google Scholar] [CrossRef] [PubMed]
- Ajellal, N.; Carpentier, J.F.; Guillaume, C.; Guillaume, S.M.; Heloua, M.; Poiriera, V.; Sarazina, Y.; Trifonovb, A. Metal-catalyzed immortal ring-opening polymerization of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef] [PubMed]
- Platel, R.H.; White, A.J.P.; Williams, C.K. Bis(phosphinic)diamido yttrium amide, alkoxide, and aryloxide complexes: An evaluation of lactide ring-opening polymerization initiator efficiency. Inorg. Chem. 2011, 50, 7718–7728. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, T.L.; Chen, C.T.; Lin, C.C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Wheaton, C.A.; Hayes, P.G.; Ireland, B.J. Complexes of Mg, Ca and Zn as homogeneous catalysts for lactide polymerization. Dalton Trans. 2009, 4832–4846. [Google Scholar] [CrossRef] [PubMed]
- Amgoune, A.; Thomas, A.M.; Carpentier, J.F. Controlled rong-opening polymerization of lactide by group 3 metal complexes. Pure Appl. Chem. 2007, 79, 2013–2030. [Google Scholar] [CrossRef]
- Sauer, A.; Kapelski, A.; Fliedel, C.; Dagorne, S.; Kol, M.; Okuda, J. Structurally well-defined group 4 metal complexesas initiators for ring-opening polymerization of lactide monomers. Dalton Trans. 2013, 42, 9007–9023. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, E. Recent advances on tailor-made titanium catalysts for biopolymer synthesis. Coord. Chem. Rev. 2016, 306, 65–85. [Google Scholar] [CrossRef]
- Dagorne, S.; Normand, M.; Kirillov, E.; Carpentier, J.F. Gallium and Indium complexes for ring-opening polymerization of cyclic ethers, esters and carbonates. Coord. Chem. Rev. 2013, 257, 1869–1886. [Google Scholar] [CrossRef] [Green Version]
- Connor, E.F.; Nyce, G.W.; Myers, M.; Möck, K.; Hedrick, J.L. First example of N-heterocyclic carbenes as catalysts for living polymerization: Organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002, 124, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, M.K.; Shin, E.J.; Hedrick, J.L.; Waymouth, R.M. Organocatalysis: Opportunities and challenges for polymer synthesis. Macromolecules 2010, 43, 2093–2107. [Google Scholar] [CrossRef]
- Acharya, A.K.; Chang, Y.A.; Jones, G.O.; Rice, J.E.; Hedrick, J.L.; Horn, H.W.; Waymouth, R.M. Experimental and computational studies on the mechanism of zwitterionic ring-opening polymerization of δ-valerolactone with N-heterocyclic carbenes. J. Phys. Chem. B 2014, 118, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Suriano, F.; Coulembier, O.; Hedrick, J.L.; Dubois, P. Functionalized cyclic carbonates: From synthesis and metal-free catalyzed ring-opening polymerization to applications. Polym. Chem. 2011, 2, 528–533. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Amgoune, A.; Bourissou, D. Dual catalysis: New approaches for the polymerization of lactones and polar olefins. Dalton Trans. 2013, 42, 9024–9029. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhang, H. Combining transition metal catalysis and organocatalysis: A broad new concept for catalysis. Chem. Soc. Rev. 2009, 40, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Shi, X. When organocatalysis meets transition-metal catalysis. Eur. J. Org. Chem. 2010, 16, 2999–3025. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Brignou, P.; Amgoune, A.; Guillaume, S.M.; Carpentier, J.F.; Bourissou, D. A dual organic/organometallic approach for catalytic ring-opening polymerization. Chem. Commun. 2011, 47, 9828–9830. [Google Scholar] [CrossRef] [PubMed]
- Brignou, P.; Guillaume, S.M.; Roisnel, T.; Bourissou, D.; Carpentier, J.-F. Discrete cationic zinc and magnesium complexes for dual organic/organometallic-catalyzed ring-opening polymerization of trimethylene carbonate. Chem. Eur. J. 2012, 18, 9360–9370. [Google Scholar] [CrossRef] [PubMed]
- Schnee, G.; Fliedel, C.; Aviles, T.; Dagrone, S. Neutral and cationic N-heterocyclic carbene zinc adducts and the BnOH/Zn(C6F5)2 binary mixture characterization and use in the ring-opening polymerization of β-Butyrolactone, lactides and trimethylene carbonate. Eur. J. Inorg. Chem. 2013, 3699–3709. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Ladaviere, C.; Amgoune, A.; Bourissou, D. Ring-opening polymerization with Zn(C6F5)2-based lewis pairs: Original and efficient approach to cyclic polyesters. J. Am. Chem. Soc. 2013, 135, 13306–13309. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Liddle, S.T.; Mungur, S.A.; Rodden, M.; Blake, A.J.; Arnold, P.L. Bifunctional yttrium(III) and titanium(IV) NHC catalysts for lactide polymerization. Chem. Commun. 2006, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Vieira, I.S.; Whitelaw, E.L.; Jones, M.D.; Pawlis, S.H. Synergistic empirical and theoretical study on the stereoselective mechanism for the aluminum salalen complex mediated polymerization of rac-lactide. Chem. Eur. J. 2013, 19, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- Dyer, H.E.; Huijser, S.; Susperregui, N.; Bonnet, F.; Schwarz, A.D.; Duchateau, R.; Maron, L.; Mountford, P. Ring-opening polymerization of rac-Lactide by bis(phenolate)amine-supported samarium borohydride complexes: An experimental and DFT Study. Organometallics 2010, 29, 3602–3621. [Google Scholar] [CrossRef]
- Broderick, E.M.; Guo, N.; Wu, T.; Vogel, C.S.; Xu, C.; Sutter, J.; Miller, J.T.; Meyer, K.; Cantat, T.; Diaconescu, P.L. Redox control of a polymerization catalyst by changing the oxidation state of the metal center. Chem. Commun. 2011, 47, 9897–9899. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Walshe, A.; Maron, L.; Baker, R.J. Ring-opening polymerization of epoxides catalyzed by uranyl complexes: An experimental and theoretical study of the reaction mechanism. Inorg. Chem. 2012, 51, 9132–9140. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Tschan, M.J.-L.; Roisnel, T.; Trivelli, X.; Gauvin, R.M.; Thomas, C.M.; Maron, L. Yttrium catalysts for syndioselective b-butyrolactone polymerization: On the origin of ligand-induced stereoselectivity. Polym. Chem. 2013, 4, 360–367. [Google Scholar] [CrossRef]
- Rosal, I.D.; Brignou, P.; Guillaume, S.M.; Carpentier, J.F.; Maron, L. DFT investigations on the ring-opening polymerization of substituted cyclic carbonates catalyzed by zinc-{β-diketiminate} complexes. Polym. Chem. 2015, 6, 3336–3352. [Google Scholar] [CrossRef] [Green Version]
- Tabthong, S.; Nanok, T.; Sumrit, P.; Kongsaeree, P.; Prabpai, S.; Chuawong, P.; Hormnirun, P. Bis(pyrrolidene) schiff base aluminum complexes as isoselective-biased initiators for the controlled ring-opening polymerization of rac-lactide: Experimental and theoretical Studies. Macromolecules 2015, 48, 6846–6861. [Google Scholar] [CrossRef]
- Marshall, E.L.; Gibson, V.C.; Rzepa, H.S. A computational analysis of the ring-opening polymerization of rac-lactide initiated by single-site β-diketiminate metal complexes: Defining the mechanistic pathway and the origin of stereocontrol. J. Am. Chem. Soc. 2005, 127, 6048–6051. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yu, I.; Mehrkhodavandi, P.; Maron, L. Theoretical investigation of lactide ring-opening polymerization induced by a dinuclear indium catalyst. Organometallics 2013, 32, 6950–6956. [Google Scholar] [CrossRef]
- Fliedel, C.; Vila-Vicosa, D.; Calhorda, M.J.; Dagorne, S.; Aviles, T. Dinuclear Zinc–N-Heterocyclic carbine complexes either the controlled ring-opening polymerization of lactide or the controlled degradation of polylactide under mild conditions. ChemCatChem 2014, 6, 1357–1367. [Google Scholar] [CrossRef]
- Xu, T.Q.; Yang, G.W.; Liu, C.; Lu, X.B. Highly robust yttrium bis(phenolate) ether catalysts for excellent isoselective ring-opening polymerization of racemic lactide. Macromolecules 2017, 50, 515–522. [Google Scholar] [CrossRef]
- Kang, X.H.; Song, Y.M.; Luo, Y.; Li, G.; Hou, Z.M.; Qu, J.P. Computational studies on isospecific polymerization of 1-hexene catalyzed by cationic rare-earth metal alkyl complex bearing a C3 iPr-trisox ligand. Macromolecules 2012, 45, 640–651. [Google Scholar] [CrossRef]
- Kang, X.H.; Yamamoto, A.; Nishiura, M.; Luo, Y.; Hou, Z.M. Computational analyses of the effect of lewis bases on styrene polymerization catalyzed by cationic scandium half-sandwich complexes. Organometallics 2015, 34, 5540–5548. [Google Scholar] [CrossRef]
- Kang, X.H.; Zhou, G.L.; Wang, X.B.; Luo, Y.; Hou, Z.M.; Qu, J.P. Alkyl effects on the chain initiation efficiency of olefin polymerization by cationic half-sandwich scandium catalysts: A DFT study. Organometallics 2016, 35, 913–920. [Google Scholar] [CrossRef]
- Kang, X.H.; Luo, Y.; Zhou, G.L.; Wang, X.B.; Yu, X.R.; Hou, Z.M.; Qu, J.P. Theoretical mechanistic studies on the trans-1,4-specific polymerization of isoprene catalyzed by a cationic La–Al binuclear complex. Macromolecules 2014, 47, 4596–4606. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, Y.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Yang, Y.; Weaver, M.N.; Merz, K.M., Jr. Assessment of the “6-31 + G** + LANL2DZ” mixed basis set coupled with density functional theory methods and the effective core potential: Prediction of heats of formation and ionization potentials for first-row-transition-metal complexes. J. Phys. Chem. A 2009, 113, 9843–9851. [Google Scholar] [CrossRef] [PubMed]
- Hoellwarth, A.; Boehme, M.; Dapprich, S.; Ehlers, A.W.; Gobbi, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al–Bi and f-type polarization functions for Zn, Cd, Hg. Chem. Phys. Lett. 1993, 208, 237–240. [Google Scholar]
- Liu, Y.; Liu, Y.; Drew, M.G.B. Correlation between regioselectivity and site charge in propene polymerisation catalysed by metallocene. Struct. Chem. 2010, 21, 21–28. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, S.; Zhang, L.; Li, H.Y.; Wang, Z.X. DFT mechanistic study of the H2-assisted chain transfer copolymerization of propylene and p-methylstyrene catalyzed by zirconocene complex. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 576–585. [Google Scholar] [CrossRef]
- Valente, A.; Zinck, P.; Mortreux, A.; Visseauxa, M.; Mendes, P.J.G.; Silva, T.J.L.; Garcia, M.H. Polymerization of ε-caprolactone using ruthenium(II) mixed metallocene catalysts and isopropyl alcohol: Living character and mechanistic study. J. Mol. Catal. A Chem. 2011, 346, 102–110. [Google Scholar] [CrossRef]
- Jitonnom, J.; Molloy, R.; Punyodom, W.; Meelua, W. Theoretical studies on aluminum trialkoxide-initiated lactone ring-opening polymerizations: Roles of alkoxide substituent and monomer ring structure. Comput. Theor. Chem. 2016, 1097, 25–32. [Google Scholar] [CrossRef]
- Jitonnom, J.; Meelua, W. Effects of silicon-bridge and π-ligands on the electronic structures and related properties of dimethyl zirconocene polymerization catalysts: A comparative theoretical study. Chiang Mai J. Sci. 2014, 41, 1220–1229. [Google Scholar]
- Jitonnom, J.; Sontag, C. Catalytic oxidation of glucose with hydrogen peroxide and colloidal gold as pseudo-homogenous catalyst: A combined experimental and theoretical investigation. Chiang Mai J. Sci. 2016, 43, 825–833. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Legault, C.Y. CLYview, Version 1.0b; University of California: Los Angeles, CA, USA, 2007. [Google Scholar]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, X.; Wei, N.N.; Hao, C.; Zhu, X.D.; He, G.H. DFT study on 1,7-octadiene polymerization catalyzed by non-bridged half-titanocene system. RSC Adv. 2016, 6, 69939–69946. [Google Scholar] [CrossRef]
- Li, Y.; Qi, X.; Lei, Y.; Lan, Y. Mechanism and selectivity for zinc-mediated cycloaddition of azides with alkynes: A computational study. RSC Adv. 2015, 5, 49802–49808. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Nijamudheen, A.; Datta, A. Mechanistic insights into the synergistic catalysis by Au(I), Ga(III), and counterions in the nakamura reaction. Org. Biomol. Chem. 2015, 13, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Luo, G.; Hou, Z.M.; Luo, Y. Mechanistic insights into scandium-catalyzed hydroaminoalkylation of olefins with amines: Origin of regioselectivity and charge-based prediction model. Organometallics 2017, 36, 1557–1565. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
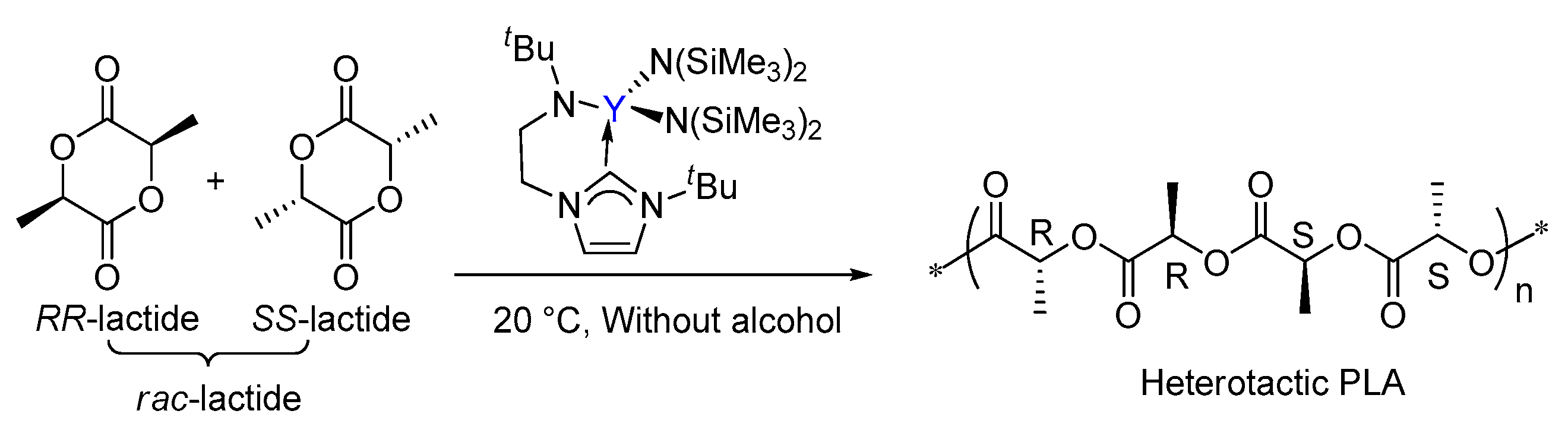{kind=link}
{kind=link}
{kind=link}
| Structure | ΔEint | ΔEdef(A) 1 | ΔEdef(B) 2 | ΔEdef | ΔE5b |
|---|---|---|---|---|---|
| 5b (RR,RR) | −26.9 | 0.9 | 19 | 19.9 | −7 |
| 5b (RR,SS) | −32.1 | 1.2 | 18.1 | 19.3 | −12.8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Mehmood, A.; Zhao, Y.; Qu, J.; Luo, Y. Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst. Inorganics 2017, 5, 46. https://doi.org/10.3390/inorganics5030046
Wang Y, Mehmood A, Zhao Y, Qu J, Luo Y. Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst. Inorganics. 2017; 5(3):46. https://doi.org/10.3390/inorganics5030046
Chicago/Turabian StyleWang, Yincheng, Andleeb Mehmood, Yanan Zhao, Jingping Qu, and Yi Luo. 2017. "Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst" Inorganics 5, no. 3: 46. https://doi.org/10.3390/inorganics5030046
APA StyleWang, Y., Mehmood, A., Zhao, Y., Qu, J., & Luo, Y. (2017). Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst. Inorganics, 5(3), 46. https://doi.org/10.3390/inorganics5030046