Synthesis and Reactivity of Mn–CF3 Complexes

Abstract
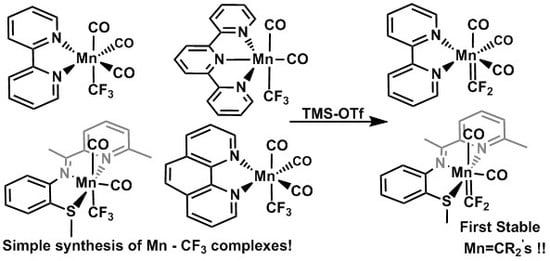
:
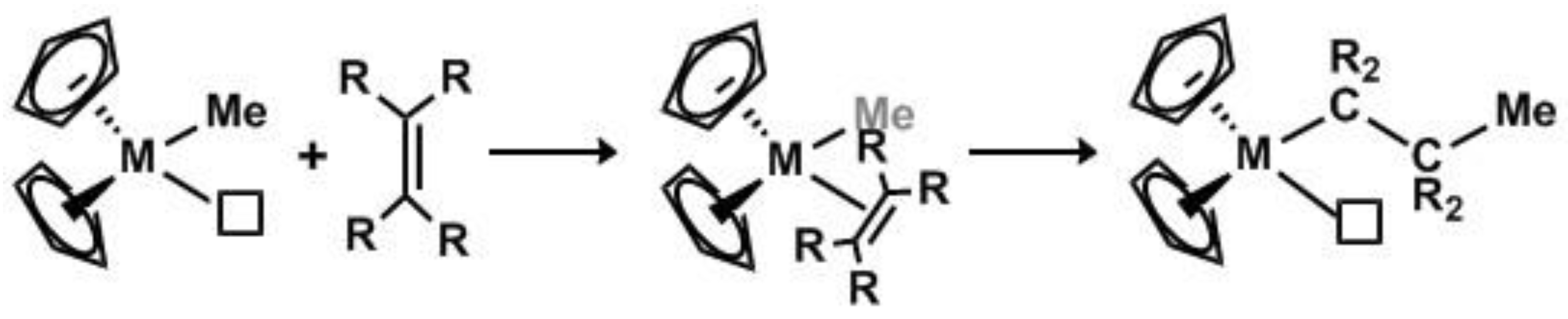
1. Introduction
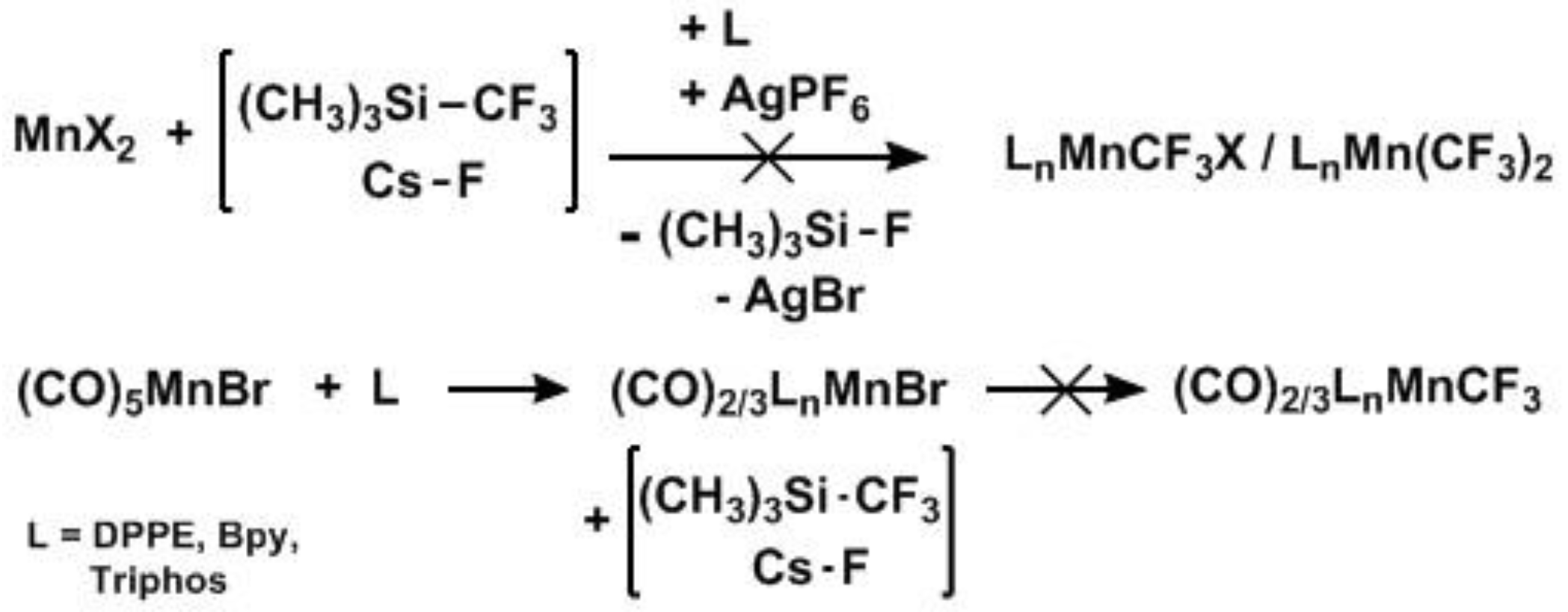
2. Results
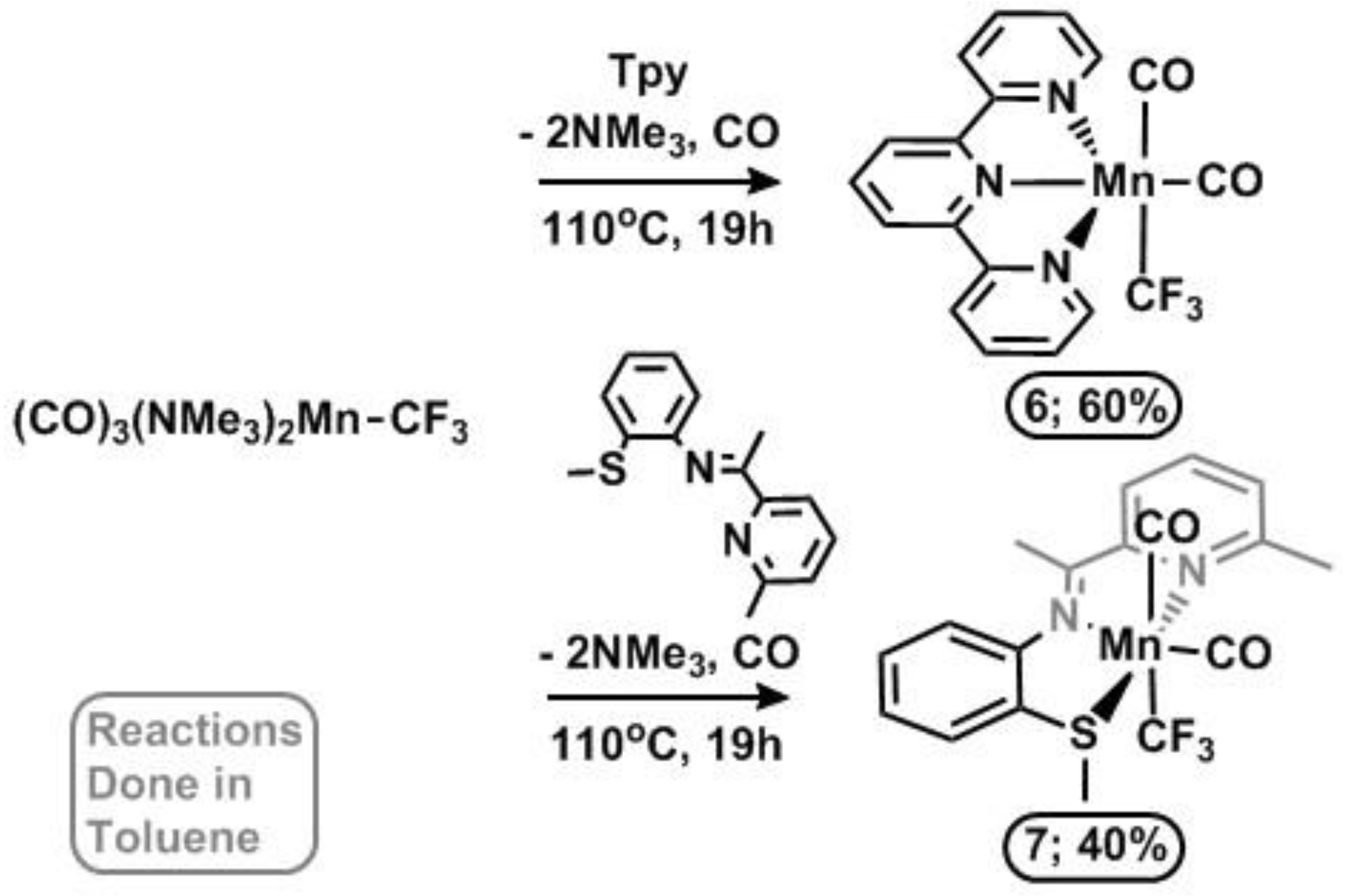
2.1. Substitution Reactions of Mn(CO)5COCF3 to form New Mn(I)–CF3 Complexes
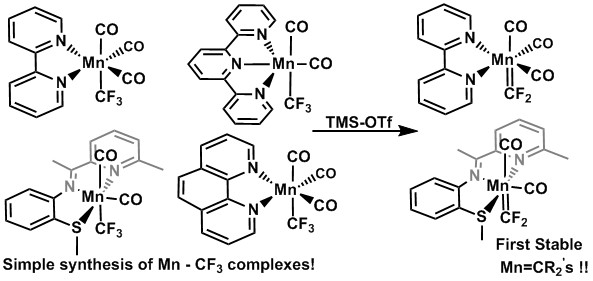
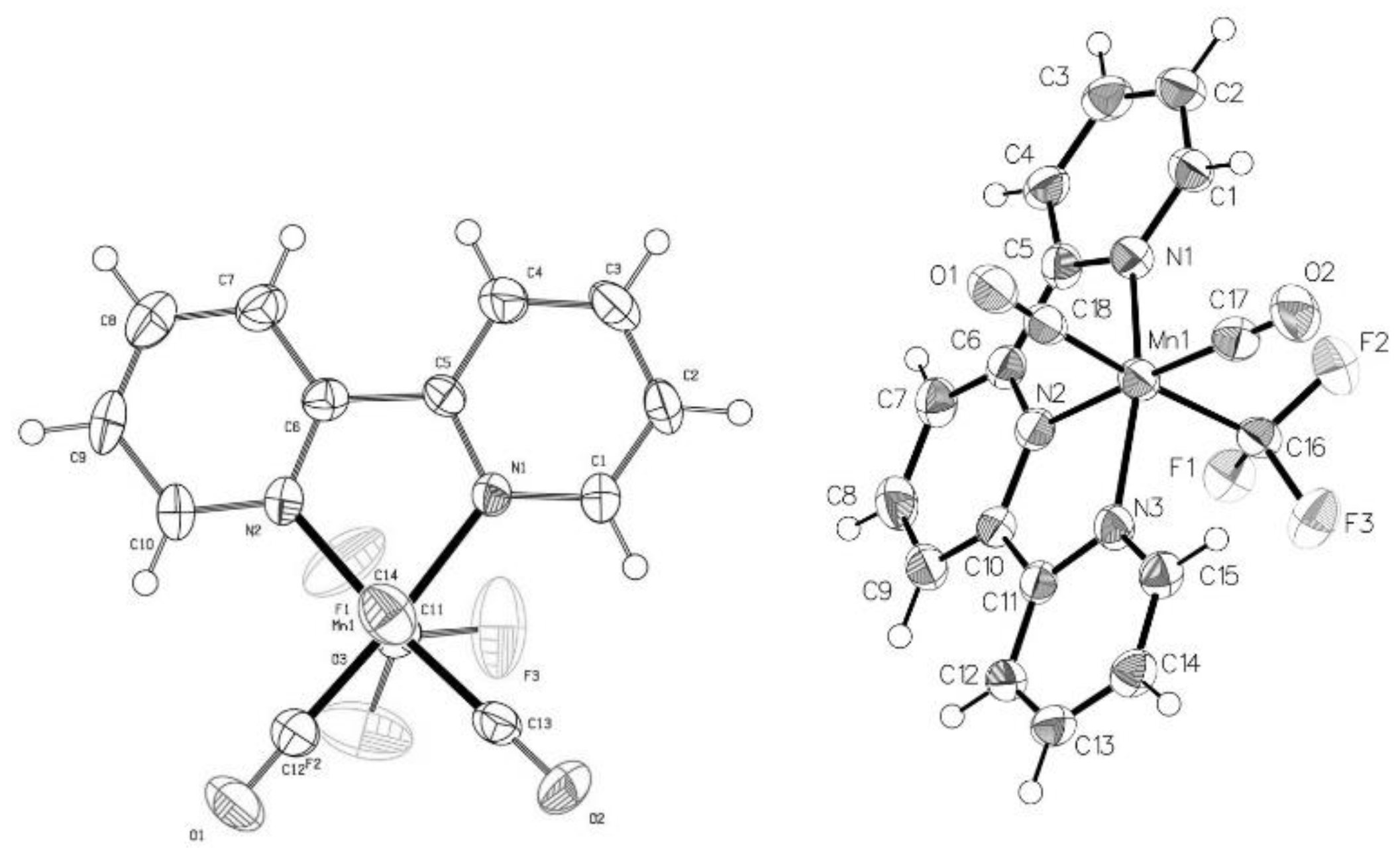
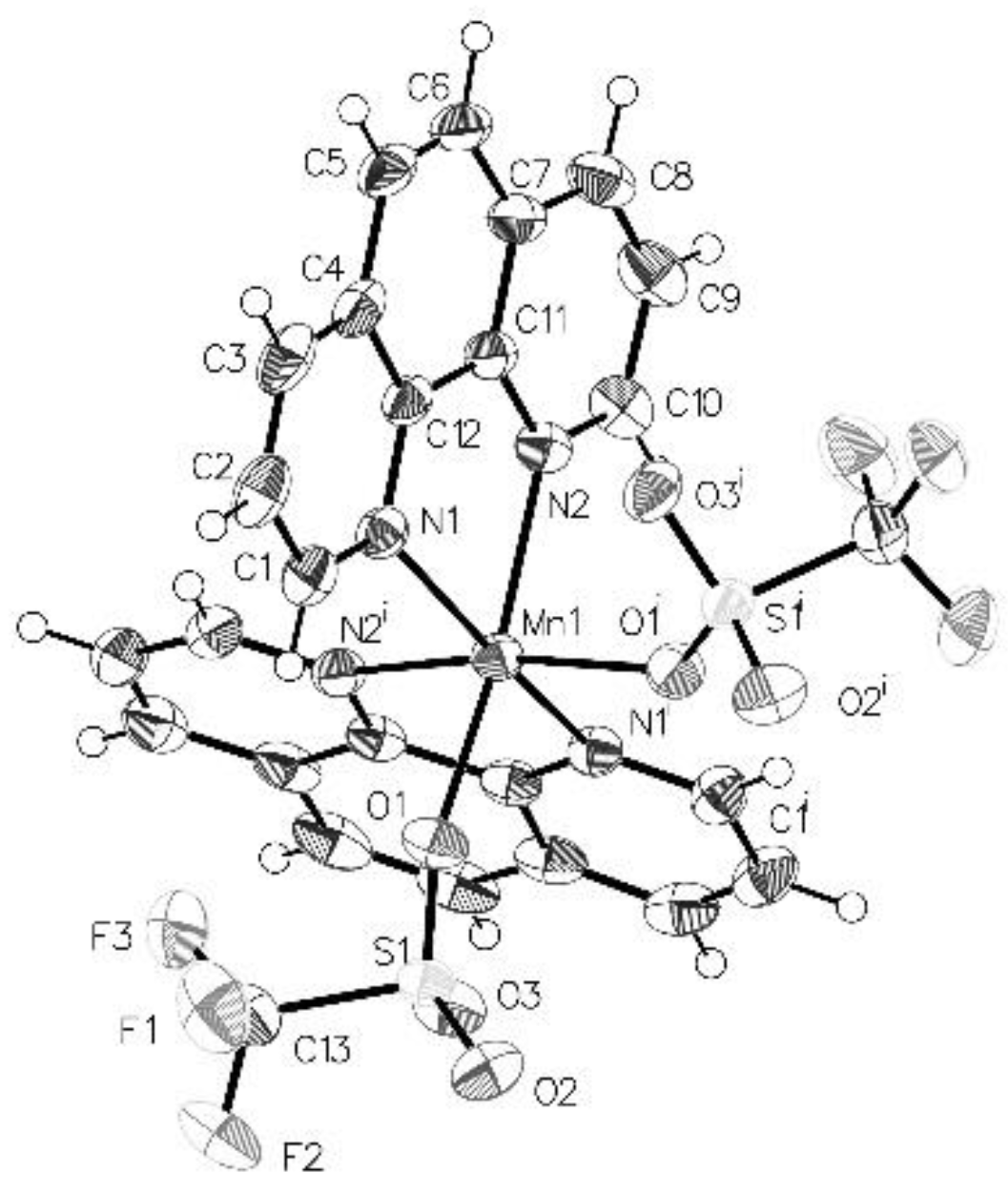
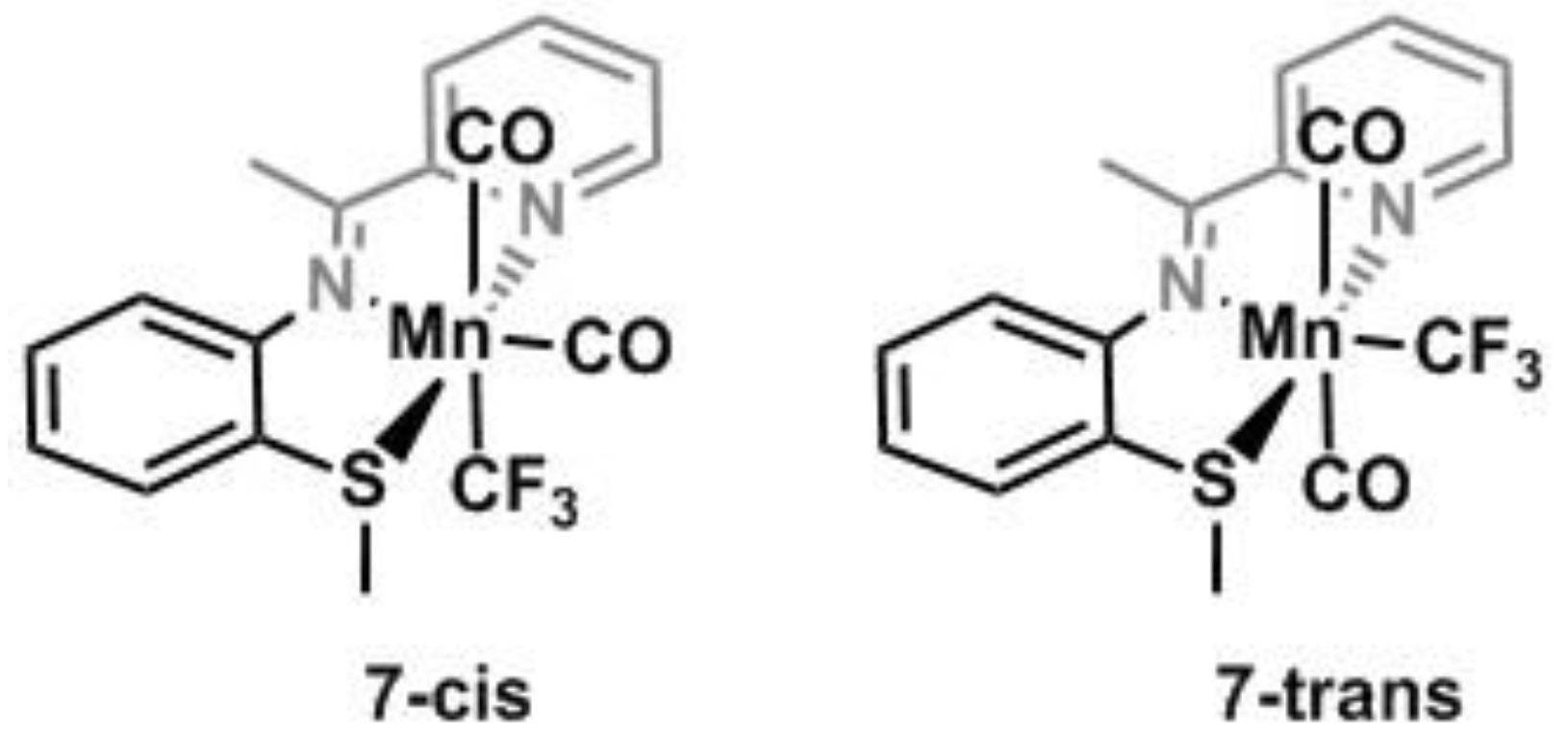
2.2. Solid State Structures of New Mn–CF3 Complexes
2.3. NMR Data for Mn–CF3
2.4. IR data
2.5. Cyclic Voltammetry Data
2.6. Mass Spectrometry
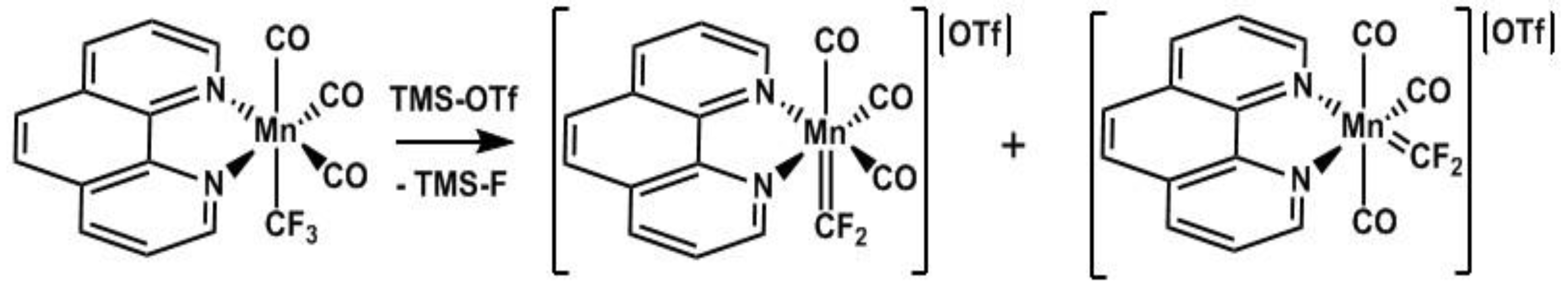
2.7. Reactivity
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steinborn, D. Fundamentals of Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Chiusoli, G.P.; Maitlis, P.M. (Eds.) Metal-Catalysis in Industrial Organic Processes; RSC Publishing: Cambridge, UK, 2006. [Google Scholar]
- Taw, F.L.; Clark, A.E.; Mueller, A.H.; Janicke, M.T.; Cantat, T.; Scott, B.L.; Hay, P.J.; Hughes, R.P.; Kiplinger, J.L. Titanium(IV) Trifluoromethyl Complexes: New Perspectives on Bonding from Organometallic Fluorocarbon Chemistry. Organometallics 2012, 31, 1484–1499. [Google Scholar] [CrossRef]
- Hughes, R.P. Adv. Organo-transition Metal Compounds Containing Perfluorinated Ligands. Organomet. Chem. 1990, 31, 183–267. [Google Scholar]
- Brothers, P.J.; Roper, W.R. Transition-metal Dihalocarbene Complexes. Chem. Rev. 1988, 88, 1293–1326. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Blackwell: Chichester, UK, 2009. [Google Scholar]
- Chambers, R.D. Fluorine in Organic Chemistry; Blackwell: Oxford, UK, 2004. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Alonso, C.; de Marigorta, E.M.; Rubiales, G.; Palacios, F. Carbon Trifluoromethylation Reactions of Hydrocarbon Derivatives and Heteroarenes. Chem. Rev. 2015, 115, 1847–1935. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent Advances in Trifluoromethylation Methodology and new CF3-containing Drugs. J. Fluor. Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Jeschke, P. The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Im, J.; Walshe-Langford, G.E.; Moon, J.-W.; Löffler, F.E. Environmental Fate of the Next Generation Refrigerant 2,3,3,3-Tetrafluoropropene (HFO-1234yf). Environ. Sci. Technol. 2014, 48, 13181–13187. [Google Scholar] [CrossRef] [PubMed]
- Panferova, L.I.; Miloserdov, F.M.; Lishchynskyi, A.; Martinez Belmonte, M.; Benet-Buchholz, J.; Grushin, V.V. Well-Defined CuC2F5 Complexes and Pentafluoroethylation of Acid Chlorides. Angew. Chem. Int. Ed. 2015, 54, 5218–5222. [Google Scholar] [CrossRef]
- Konovalov, A.I.; Lishchynskyi, A.; Grushin, V.V. Mechanism of Trifluoromethylation of Aryl Halides with CuCF3 and the Ortho Effect. J. Am. Chem. Soc. 2014, 136, 13410–13425. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Tsubogo, T.; Litvinas, N.D.; Hartwig, J.F. A Broadly Applicable Copper Reagent for Trifluoromethylations and Perfluoroalkylations of Aryl Iodides and Bromides. Angew. Chem. Int. Ed. 2011, 50, 3793–3798. [Google Scholar] [CrossRef] [Green Version]
- Dubinina, G.G.; Furutachi, H.; Vicic, D.A. Active Trifluoromethylating Agents from Well-Defined Copper(I)–CF3 Complexes. J. Am. Chem. Soc. 2008, 130, 8600–8601. [Google Scholar] [CrossRef] [PubMed]
- Wiemers, D.M.; Burton, D.J. Pregeneration, Spectroscopic Detection and Chemical Reactivity of (Trifluoromethyl)copper, an Elusive and Complex Species. J. Am. Chem. Soc. 1986, 108, 832–834. [Google Scholar] [CrossRef]
- McLoughlin, V.C.R.; Thrower, J. Route to Fluoroalkyl-substituted Aromatic Compounds Involving Fluoroalkylcopper Intermediates. Tetrahedron 1969, 25, 5921–5940. [Google Scholar] [CrossRef]
- Choi, W.J.; Choi, S.; Ohkubo, K.; Fukuzumi, S.; Cho, E.J.; You, Y. Mechanisms and Applications of Cyclometalated Pt(II) Complexes in Photoredox Catalytic Trifluoromethylation. Chem. Sci. 2015, 6, 1454–1464. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of Arenes and Heteroarenes by Means of Photoredox Catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Natta, G. New Class of Polymers of α-olefin Having Exceptional Regularity of Structure. Atti. Acc. Naz. Lincei. 1955, 4, 61. [Google Scholar]
- Ziegler, K.; Holzkamp, E.; Breil, H. Polymerisation von Äthylen und anderen Olefinen. Angew. Chem. 1955, 67, 541. [Google Scholar]
- Natta, G.; Pino, P.; Corradini, P.; Danusso, F.; Mantica, E.; Mazzanti, G.; Moraglio, G. Crystalline High Polymers of α-Olefins. J. Am. Chem. Soc. 1955, 77, 1708–1710. [Google Scholar] [CrossRef]
- Boday, D.J. The State of Fluoropolymers. In Advances in Fluorine-Containing Polymers; Smith, D.W., Jr., Iacono, S.T., Boday, D.J., Kettwich, S.C., Eds.; American Chemical Society: Washington, DC, USA, 2012; pp. 1–5. [Google Scholar]
- Kim, C.U.; Lee, J.M.; Ihm, S.K. Emulsion polymerization of tetrafluoroethylene: Effects of reaction conditions on particle formation. J. Fluor. Chem. 1999, 96, 11–21. [Google Scholar] [CrossRef]
- Kim, C.U.; Lee, J.M.; Ihm, S.K. Emulsion polymerization of tetrafluoroethylene: Effects of reaction conditions on the polymerization rate and polymer molecular weight. J. Appl. Polym. Sci. 1999, 73, 777–793. [Google Scholar] [CrossRef]
- Sianesi, D.; Caporiccio, G. Stereospecific Polymerization of Perfluoroolefins. Makromol. Chem. 1963, 60, 213–222. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Zhou, J.; Zhu, Z.; Hou, G. π-Bis(Benzene)Chromium(0)-Catalyzed Oligomerization of Perfluoropropylene. J. Organomet. Chem. 1981, 205, 185–191. [Google Scholar] [CrossRef]
- Clot, E.; Megret, C.; Kraft, B.M.; Eisennstein, O.; Jones, W.D. Defluorination of Perfluoropropene Using Cp*2ZrH2 and Cp*2ZrHF: A Mechanism Investigation from a Joint Experimental-Theoretical Perspective. J. Am. Chem. Soc. 2004, 126, 5647–5653. [Google Scholar] [CrossRef] [PubMed]
- Smart, B.E. The Chemistry of Functional Groups, Supplement D; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 1983; Chapter 14. [Google Scholar]
- Wilford, J.B.; Stone, F.G.A. Chemistry of the Metal Carbonyls. XXVIII. Addition of Rhenium Pentacarhonyl Hydride to Fluoro Olefins. Inorg. Chem. 1965, 4, 93–97. [Google Scholar] [CrossRef]
- Fujisawa, K.; Nabika, M. Development of New Polymerization Catalysts with Manganese(II) Complexes. Coord. Chem. Rev. 2013, 257, 119–129. [Google Scholar] [CrossRef]
- Algarra, A.G.; Grushin, V.V.; Macgregor, S.A. Natural Bond Orbital Analysis of the Electronic Structure of [LnM(CH3)] and [LnM(CF3)] Complexes. Organometallics 2012, 31, 1467–1476. [Google Scholar] [CrossRef]
- McClellan, W.R. Perfluoroalkyl and Perfluoroacyl Metal Carbonyls. J. Am. Chem. Soc. 1961, 83, 1598–1600. [Google Scholar] [CrossRef]
- Naumann, D.; Kaiser, M. A New Synthesis of Perfluoroorgano Manganese and Rhenium Compounds. Z. Anorg. Allg. Chem. 1995, 621, 812–816. [Google Scholar] [CrossRef]
- Banerjee, S.; Karunananda, M.K.; Bagherzadeh, S.; Jayarathne, U.; Parmelee, S.R.; Waldhart, G.W.; Mankad, N.P. Synthesis and Characterization of Heterobimetallic Complexes with Direct Cu-M Bonds (M = Cr, Mn, Co, Mo, Ru, W) Supported by N-Heterocyclic Carbene Ligands: A Toolkit for Catalytic Reaction Discovery. Inorg. Chem. 2014, 53, 11307–11315. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, S.E.A.; Durgaprasad, G.; Muthiah, K.A.T.; Rose, M.J. Tuning Coordination Modes of Pyridine/thioether Schiff Base (NNS) Ligands to Mononuclear Manganese Carbonyls. Dalton Trans. 2014, 43, 10725–10738. [Google Scholar] [CrossRef] [PubMed]
- Welch, K.D.; Dougherty, W.G.; Kassel, W.S.; Dubois, D.L.; Bullock, R.M. Synthesis, Structure and Reactions of Manganese Complexes Containing Diphosphine Ligands and Pendant Amines. Organometallics 2010, 29, 4532–4540. [Google Scholar] [CrossRef]
- Van Putten, R.; Uslamin, E.A.; Garbe, M.; Liu, C.; Gonzalez-de-Castro, A.; Lutz, M.; Junge, K.; Hensen, E.J.M.; Beller, M.; Lefort, L.; et al. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem. Int. Ed. 2017, 56, 7531–7534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S. Tripodal Phosphine Ligands. Syntheses and Coordination Chemistry Toward Mn(I). J. Chin. Chem. Soc. 1992, 39, 611–616. [Google Scholar] [CrossRef]
- Luh, T.-Y. Trimethylamine N-oxide—A Versatile Reagent for Organometallic Chemistry. Coord. Chem. Rev. 1984, 60, 255–276. [Google Scholar] [CrossRef]
- Morrison, J.A. Trifluoromethyl-containing Transition Metal Complexes. Adv. Organomet. Chem. 1993, 35, 211–239. [Google Scholar]
- Leyssens, T.; Peeters, D.; Orpen, A.G.; Harvey, J.N. How Important is Metal-Ligand Back-bonding toward YX3 Ligands (Y = N, P, C, Si)? An NBO Analysis. Organometallics 2007, 26, 2637–2645. [Google Scholar] [CrossRef]
- Richmond, T.G.; Shriver, D.F. Electrophillic Halogen Exchange between Lewis Acids and Transition-Metal Perfluoroalkyl Complexes. Synthesis and Characterization of Transition Metal α-HaloAlkyl Complexes. Organometallics 1984, 3, 305–314. [Google Scholar] [CrossRef]
- Richmond, T.G.; Crespi, A.M.; Shriver, D.F. Nucleophillic, Electrophillic, and Homolytic Reaction Chemistry of Transition Metal Carbonyl Trihalomethyl (X = F, Cl, Br) Complexes. Organometallics 1984, 3, 314–319. [Google Scholar] [CrossRef]
- Harrison, D.J.; Daniels, A.L.; Guan, J.; Gabidullin, B.M.; Hall, M.B.; Baker, R.T. Nickel Fluorocarbene Metathesis with Fluoroalkenes. Angew. Chem. Int. Ed. 2018, 57, 5772–5776. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Daniels, A.L.; Korobkov, I.; Baker, R.T. d10 Nickel Difluorocarbenes and Their Cycloaddition Reactions with Tetrafluoroethylene. Organometallics 2015, 34, 5683–5686. [Google Scholar] [CrossRef]
- Harrison, D.J.; Lee, G.M.; Leclerc, M.C.; Korobkov, I.; Baker, R.T. Cobalt Fluorocarbenes: Cycloaddition Reactions with Tetrafluoroethylene and Reactivity of the Perfluorometallacyclic Products. J. Am. Chem. Soc. 2013, 135, 18296–18299. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Helquist, P. Cyclopropanation of Olefins with a Stable, Iron-containing Methylene Transfer Agent. J. Am. Chem. Soc. 1979, 101, 6473–6475. [Google Scholar] [CrossRef]
- O’Connor, E.J.; Brandt, S.; Helquist, P. (η5-C5H5)(CO)2FeCH2S+(CH3)2BF4−: A Methylene Transfer Reagent for the Direct Cyclopropanation of Alkenes. J. Am. Chem. Soc. 1987, 109, 3739–3947. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
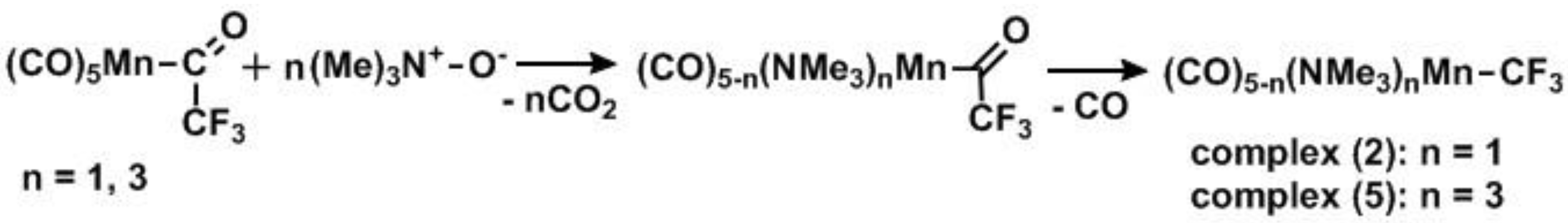{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Mn–CCF3 | Mn–CCO | Mn–N |
|---|---|---|---|
| Mn(CO)5CF3 (1) | 2.056(5) | NA | NA |
| 1.789(5) (cis) | - | ||
| Mn(CO)3(Bpy)CF3 (3) | 2.039(7) | 1.780(7) (cis) | 2.041(2) |
| 1.823(1) (trans) | 2.034(8) | ||
| Mn(CO)2(Tpy)CF3 (6) | 2.096(1) | 1.772(3) (cis) | 2.021(1), 2.025(1) |
| 1.818(9) (trans) | 1.959(2) (N2) |
| Complex | CO Stretching Frequencies (cm−1) |
|---|---|
| Mn(CO)5CF3 (1) | 2140, 2040, 2010 |
| Mn(CO)3(Bpy)CF3 (3) | 2020, 1910 |
| Mn(CO)3(Phen)CF3 (4) | 2010, 1930 |
| Mn(CO)2(Tpy)CF3 (6) | 2020, 1900, 1850 |
| Mn(CO)2(NNS)CF3 (7) | 2020, 1910, 1900 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daniels, A.L.; Da Gama, J.G.; Edjoc, R.; Gabidullin, B.M.; Baker, R.T. Synthesis and Reactivity of Mn–CF3 Complexes. Inorganics 2019, 7, 3. https://doi.org/10.3390/inorganics7010003
Daniels AL, Da Gama JG, Edjoc R, Gabidullin BM, Baker RT. Synthesis and Reactivity of Mn–CF3 Complexes. Inorganics. 2019; 7(1):3. https://doi.org/10.3390/inorganics7010003
Chicago/Turabian StyleDaniels, Alex L., Jason G. Da Gama, Racquel Edjoc, Bulat M. Gabidullin, and R. Tom Baker. 2019. "Synthesis and Reactivity of Mn–CF3 Complexes" Inorganics 7, no. 1: 3. https://doi.org/10.3390/inorganics7010003
APA StyleDaniels, A. L., Da Gama, J. G., Edjoc, R., Gabidullin, B. M., & Baker, R. T. (2019). Synthesis and Reactivity of Mn–CF3 Complexes. Inorganics, 7(1), 3. https://doi.org/10.3390/inorganics7010003