Synthesis and Structural Characterization of Two New Main Group Element Carboranylamidinates

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
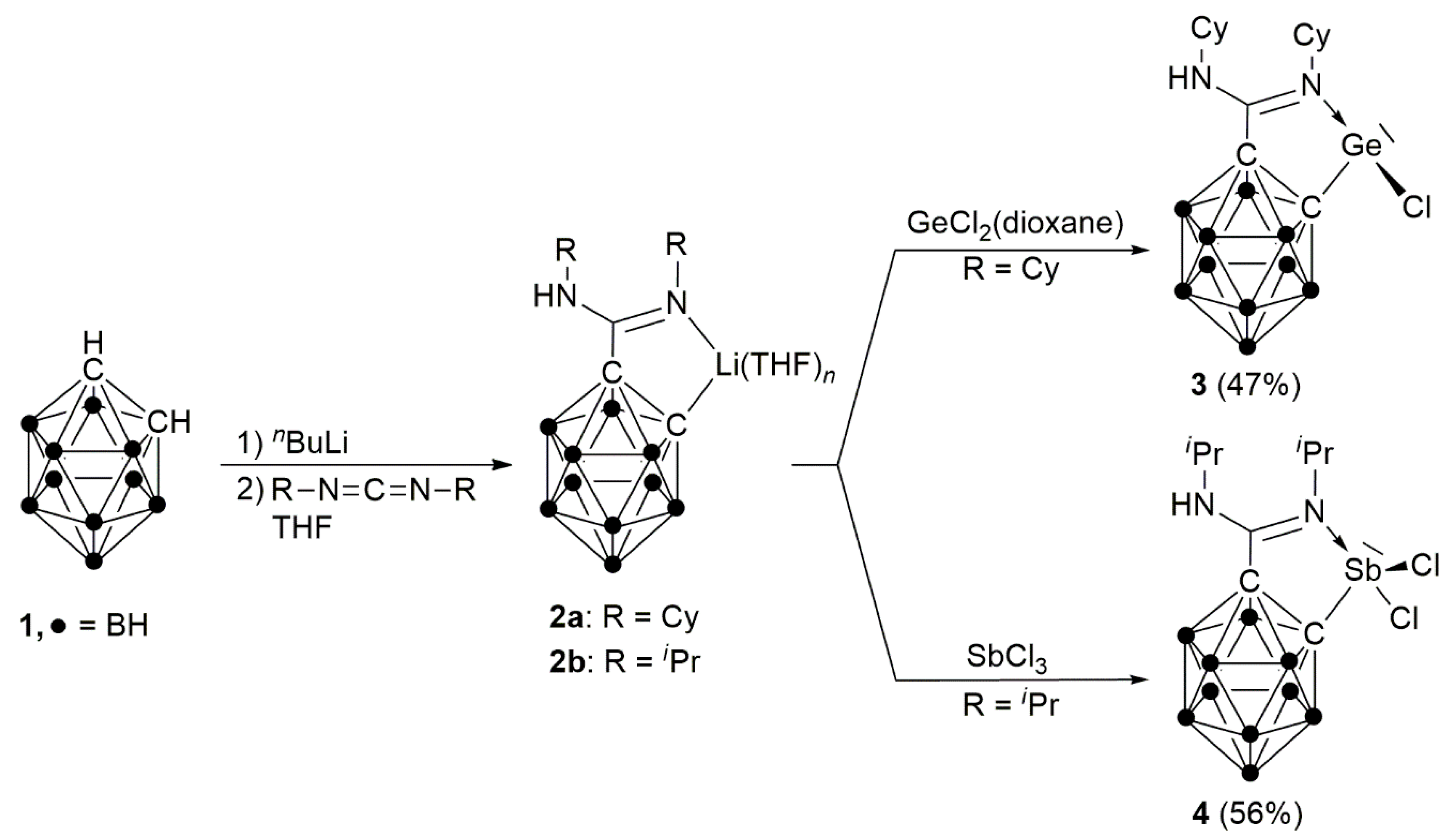
2. Results and Discussion
2.1. Synthesis and Characterization of GeCl[HLCy] (3) and SbCl2[HLiPr] (4)
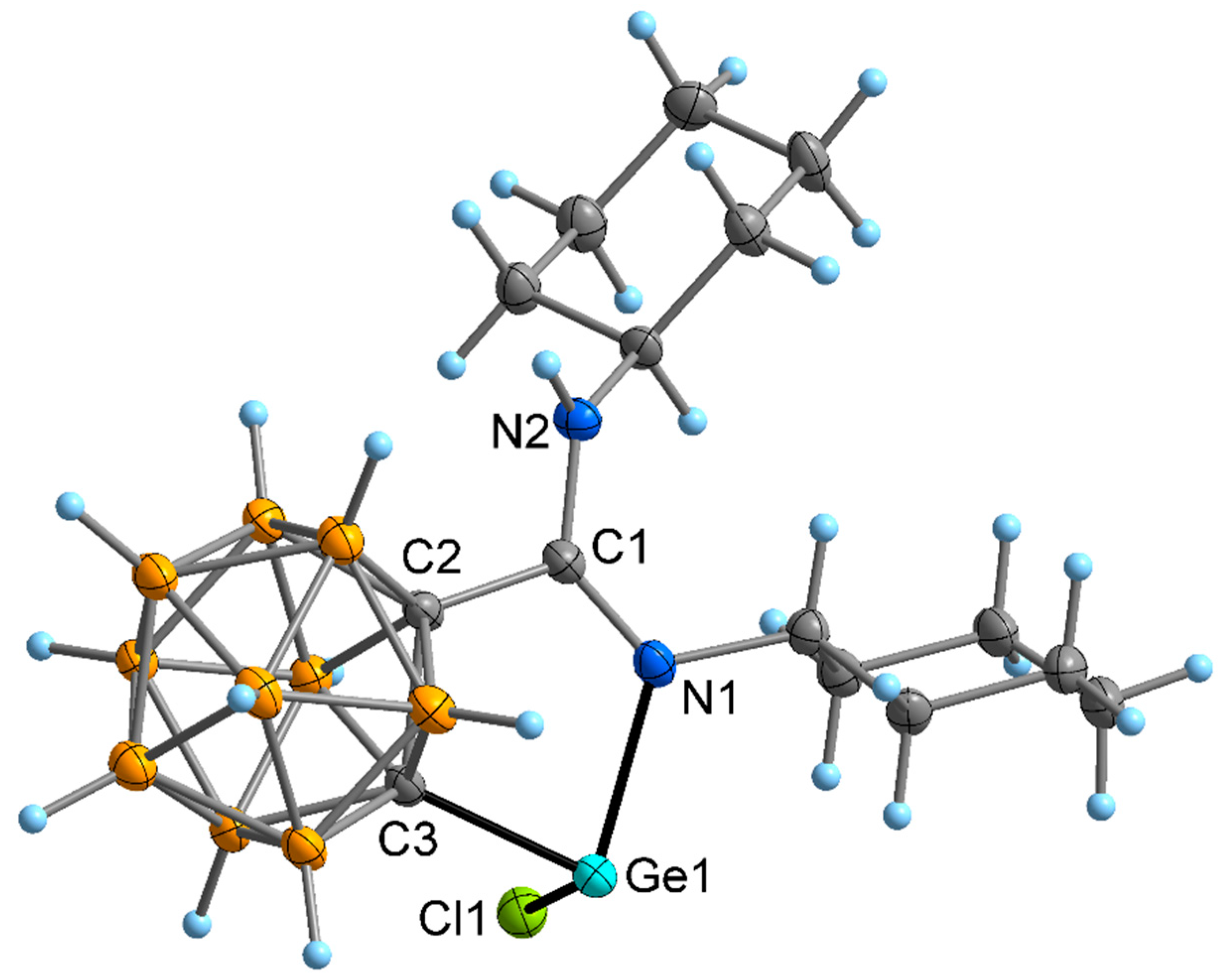
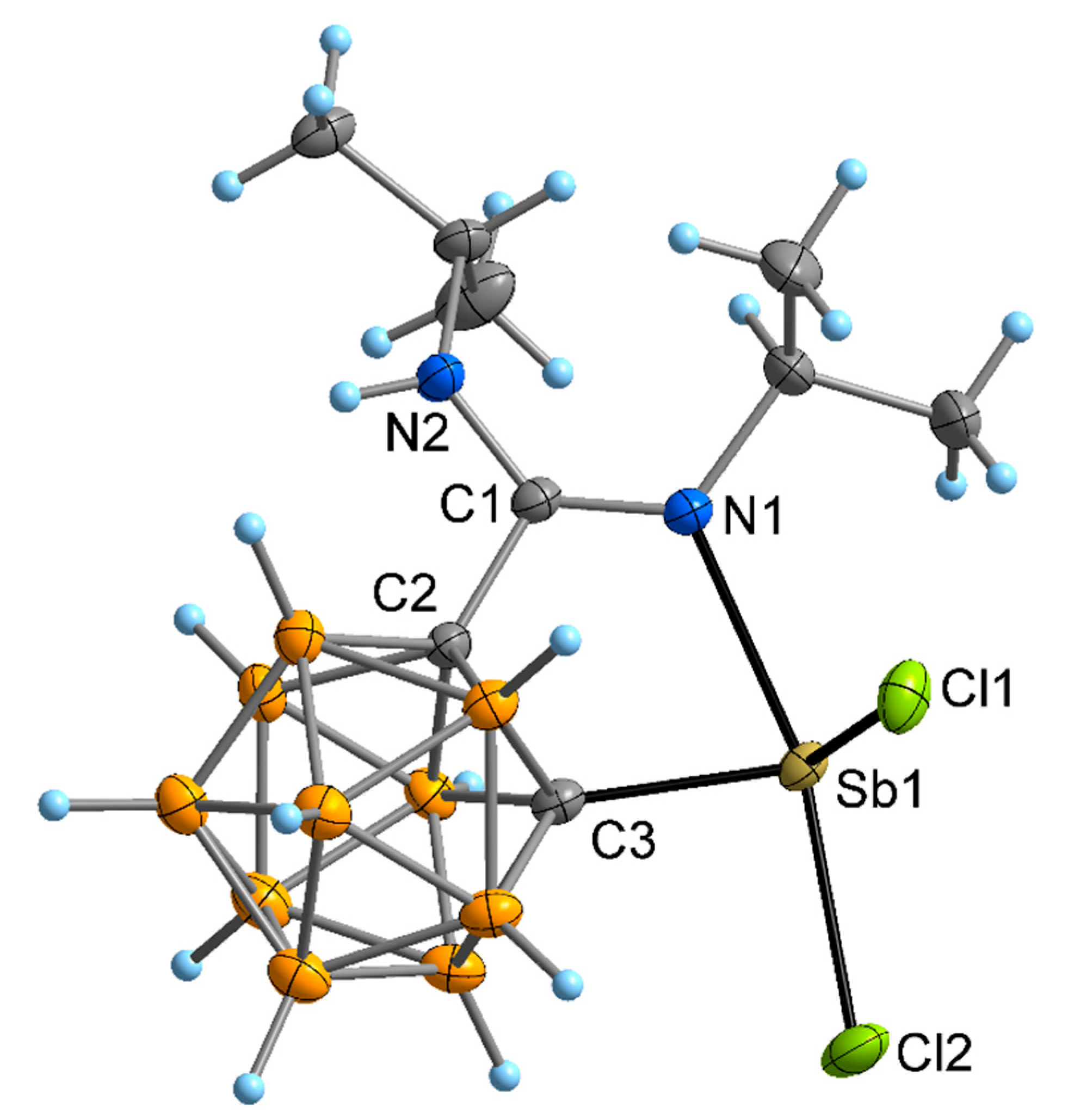
2.2. Crystal and Molecular Structures
3. Experimental Section
3.1. General Procedures and Instrumentation
3.2. Synthesis of Compound 3
3.3. Synthesis of Compound 4
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zakharkin, L.I.; Stanko, V.I.; Brattsev, V.A.; Chapovskii, Y.A.; Okhlobystin, O.Y. Synthesis of a new class of organoboron compounds, B10C2H12 (barene) and its derivatives. Russ. Chem. Bull. 1963, 12, 2074. [Google Scholar] [CrossRef]
- Brown, A.D.; Colquhoun, H.M.; Daniels, A.J.; MacBride, J.A.H.; Stephenson, I.R.; Wade, K. Polymers and ceramics based on icosahedral carboranes. Model studies of the formation and hydrolytic stability of aryl ether, ketone, amide and borane linkages between carborane units. J. Mater. Chem. 1992, 2, 793–804. [Google Scholar] [CrossRef]
- Belmont, J.A.; Soto, J.; King, R.E., III; Donaldson, A.J.; Hewes, J.D.; Hawthorne, M.F. Metallacarboranes in catalysis. 8. I: Catalytic hydrogenolysis of alkenyl acetates. II: Catalytic alkene isomerization and hydrogenation revisited. J. Am. Chem. Soc. 1989, 111, 7475–7486. [Google Scholar] [CrossRef]
- Teixidor, F.; Flores, M.A.; Viñas, C.; Kivekäs, R.; Sillanpää, R. [Rh(7-SPh-8-Me-7,8-C2B9H10)(PPh3)2]: A New Rhodacarborane with Enhanced Activity in the Hydrogenation of 1-Alkenes. Angew. Chem. Int. Ed. 1996, 35, 2251–2253. [Google Scholar] [CrossRef]
- Ferlekidis, A.; Goblet-Stachow, M.; Liégeois, J.F.; Pirotte, B.; Delarge, J.; Demonceau, A.; Fontaine, M.; Noels, A.F.; Chizhevsky, I.T.; Zinevich, T.V.; et al. Ligand effects in the hydrogenation of methacycline to doxycycline and epi-doxycycline catalysed by rhodium complexes molecular structure of the key catalyst [closo-3,3-(η2,3-C7H7CH2)-3,1,2-RhC2B9H11]. J. Organomet. Chem. 1997, 536/537, 405–412. [Google Scholar] [CrossRef]
- Vaillant, J.F.; Guenther, K.J.; King, A.S.; Morel, P.; Schaffer, P.; Sogbein, O.O.; Stephenson, K. The medicinal chemistry of carboranes. Coord. Chem. Rev. 2002, 232, 173–230. [Google Scholar] [CrossRef]
- Murophy, D.M.; Mingos, D.M.P.; Haggitt, J.L.; Poell, H.R.; Westcott, S.A.; Marder, T.B.; Taylor, N.J.; Kanis, D.R. Synthesis of icosahedral carboranes for second-harmonic generation. Part 2. J. Mater. Chem. 1993, 3, 139–148. [Google Scholar] [CrossRef]
- Dröse, P.; Hrib, C.G.; Edelmann, F.T. Carboranylamidinates. J. Am. Chem. Soc. 2010, 132, 15540–15541. [Google Scholar] [CrossRef]
- Junk, P.C.; Cole, M.L. Alkali-metal bis(aryl)formamidinates: A study of coordinative versatility. Chem. Commun. 2007, 1579–1590. [Google Scholar] [CrossRef]
- Edelmann, F.T. Chapter 3-Advances in the Coordination Chemistry of Amidinate and Guanidinate Ligands. Adv. Organomet. Chem. 2008, 57, 183–352. [Google Scholar]
- Edelmann, F.T. Lanthanide amidinates and guanidinates in catalysis and materials science: A continuing success story. Chem. Soc. Rev. 2012, 41, 7657–7672. [Google Scholar] [CrossRef] [PubMed]
- Deacon, G.B.; Hossain, M.E.; Junk, P.C.; Salehisaki, M. Rare-earth N,N’-diarylformamidinate complexes. Coord. Chem. Rev. 2017, 340, 247–265. [Google Scholar] [CrossRef]
- Yao, Z.-J.; Su, G.; Jin, G.-X. Versatile Reactivity of Half-Sandwich Ir and Rh Complexes toward Carboranylamidinates and Their Derivatives: Synthesis, Structure, and Catalytic Activity for Norbornene Polymerization. Chem. Eur. J. 2011, 17, 13298–13307. [Google Scholar] [CrossRef] [PubMed]
- Yaso, Z.-J.; Xu, B.; Su, G.; Jin, G.-X. B–H bond activation half-sandwich Ir and Ru complexes containing carboranylamidinate selenolate ligands. J. Organomet. Chem. 2012, 721–722, 31–35. [Google Scholar]
- Yalo, Z.-J.; Yue, Y.-J.; Jin, G.-X. C–C Bond Cleavage of Zwitterionic Carboranes Promoted by a Half-Sandwich Iridium(III) Compley. Chem. Eur. J. 2013, 19, 2611–2614. [Google Scholar]
- Xu, B.; Yao, Z.-J.; Jin, G.-X. Reactivity of half-sandwich metal complexes with sterically encumbered N,N′-bis(2,6-diisopropylphenyl) group-substituted carboranylamidinate ligands. Russ. Chem. Bull. 2014, 63, 963–969. [Google Scholar] [CrossRef]
- Hillebrand, P.; Hrib, C.G.; Harmgarth, N.; Jones, P.G.; Lorenz, V.; Kühling, M.; Edelmann, F.T. Carboranylamidinates of di- and trivalent iron. Inorg. Chem. Commun. 2014, 46, 127–129. [Google Scholar] [CrossRef]
- Rädisch, T.; Harmgarth, N.; Liebing, P.; Beltrán-Leiva, M.J.; Páez-Hernández, D.; Arratia-Pérez, R.; Engelhardt, F.; Hilfert, L.; Oehler, F.; Busse, S.; et al. Three new types of transition metal carboranylamidinate complexes. Dalton Trans. 2018, 47, 6666–6671. [Google Scholar] [CrossRef]
- Yao, Z.-J.; Jin, G.-X. Synthesis, Reactivity, and Structural Transformation of Mono- and Binuclear Carboranylamidinate-Based 3d Metal Complexes and Metallacarborane Derivatives. Organometallics 2012, 31, 1767–1774. [Google Scholar] [CrossRef]
- Harmgarth, N.; Gräsing, D.; Dröse, P.; Hrib, C.G.; Jones, P.G.; Lorenz, V.; Hilfert, L.; Busse, S.; Edelmann, F.T. Novel inorganic heterocycles from dimetalated carboranylamidinates. Dalton Trans. 2014, 43, 5001–5013. [Google Scholar] [CrossRef] [PubMed]
- Harmgarth, N.; Liebing, P.; Hillebrand, P.; Busse, S.; Edelmann, F.T. Synthesis and crystals structures of two new tin bis(carboranylamidinate) complexes. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, 73, 1443–1448. [Google Scholar] [CrossRef]
- Harmgarth, N.; Liebing, P.; Förster, A.; Hilfert, L.; Busse, S.; Edelmann, F.T. Spontaneous vs. base-induced dehydrochlorination of Group 14 ortho-carboranylamidinates. Eur. J. Inorg. Chem. 2017, 2017, 4473–4479. [Google Scholar] [CrossRef]
- Edelmann, F.T. Carboranylamidinates. Z. Anorg. Allg. Chem. 2013, 639, 655–667. [Google Scholar] [CrossRef]
- Yao, Z.-J.; Jin, G.-X. Transition metal complexes based on carboranyl ligands containing N, P, and S donors: Synthesis, reactivity and applications. Coord. Chem. Rev. 2013, 257, 2522–2535. [Google Scholar] [CrossRef]
- Kolesnikov, S.P.; Rogozhin, I.S.; Nefedov, O.M. Preparation of complex of germanium bichloride with 1,4-dioxane. Bull. Acad. Sci. USSR Div. Chem. Sci. 1974, 23, 2297–2298. [Google Scholar] [CrossRef]
- Groom, C.R.; Allen, F.H. The Cambridge Structural Database in retrospect and prospect. Angew. Chem. Int. Ed. 2014, 53, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Stoe & Cie. X-Area and X-Red; Stoe & Cie: Darmstadt, Germany, 2002. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structures refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liebing, P.; Harmgarth, N.; Zörner, F.; Engelhardt, F.; Hilfert, L.; Busse, S.; Edelmann, F.T. Synthesis and Structural Characterization of Two New Main Group Element Carboranylamidinates. Inorganics 2019, 7, 41. https://doi.org/10.3390/inorganics7030041
Liebing P, Harmgarth N, Zörner F, Engelhardt F, Hilfert L, Busse S, Edelmann FT. Synthesis and Structural Characterization of Two New Main Group Element Carboranylamidinates. Inorganics. 2019; 7(3):41. https://doi.org/10.3390/inorganics7030041
Chicago/Turabian StyleLiebing, Phil, Nicole Harmgarth, Florian Zörner, Felix Engelhardt, Liane Hilfert, Sabine Busse, and Frank T. Edelmann. 2019. "Synthesis and Structural Characterization of Two New Main Group Element Carboranylamidinates" Inorganics 7, no. 3: 41. https://doi.org/10.3390/inorganics7030041
APA StyleLiebing, P., Harmgarth, N., Zörner, F., Engelhardt, F., Hilfert, L., Busse, S., & Edelmann, F. T. (2019). Synthesis and Structural Characterization of Two New Main Group Element Carboranylamidinates. Inorganics, 7(3), 41. https://doi.org/10.3390/inorganics7030041