Comparing the Acidity of (R3P)2BH-Based Donor Groups in Iridium Pincer Complexes


Abstract
:
1. Introduction
2. Results and Discussion
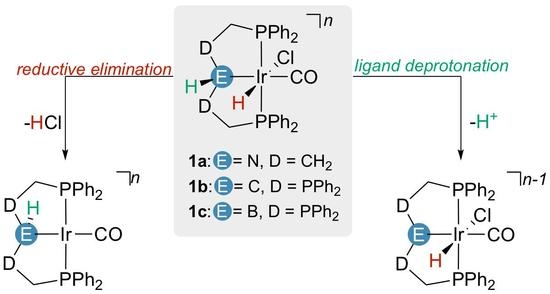
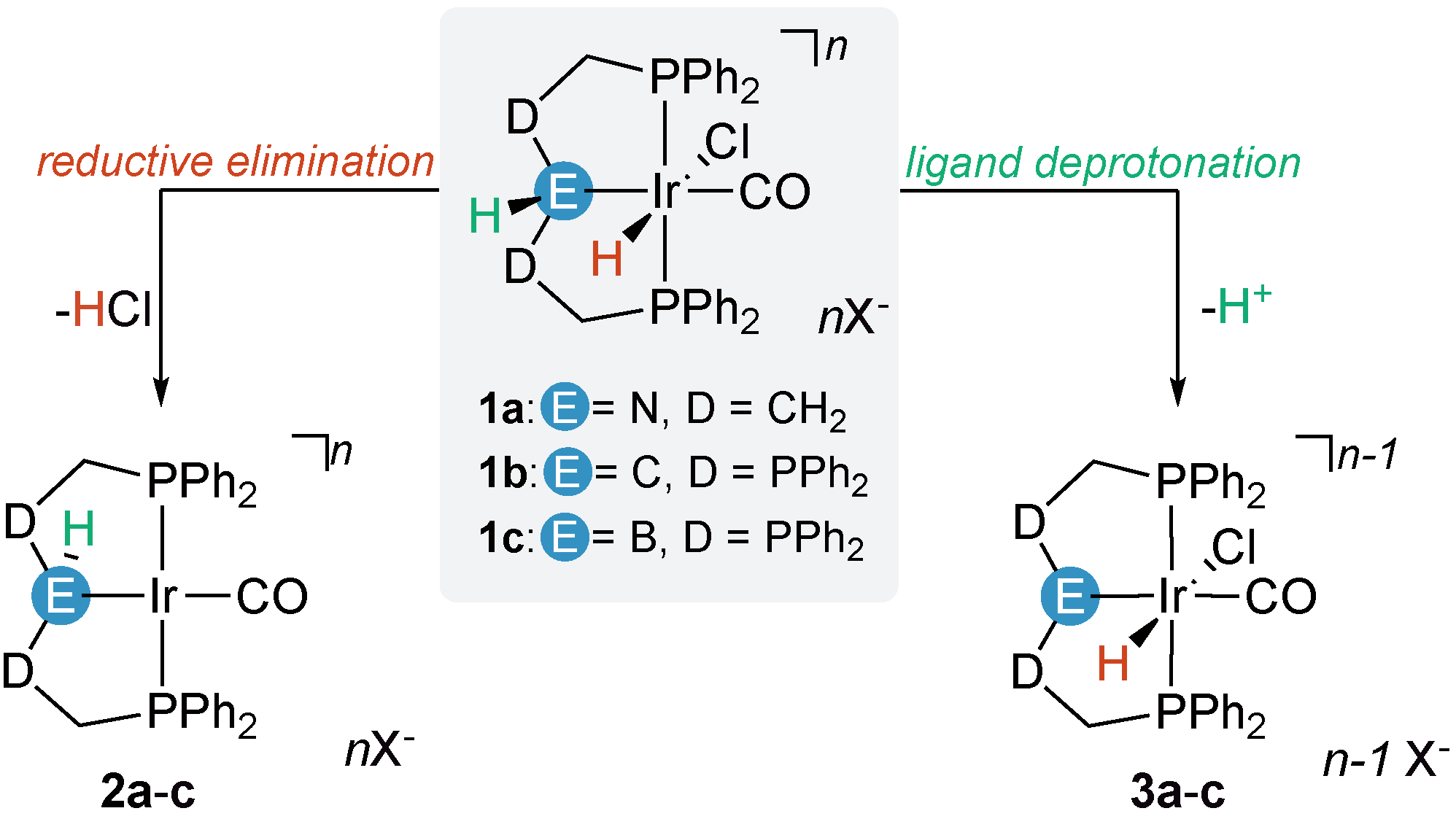
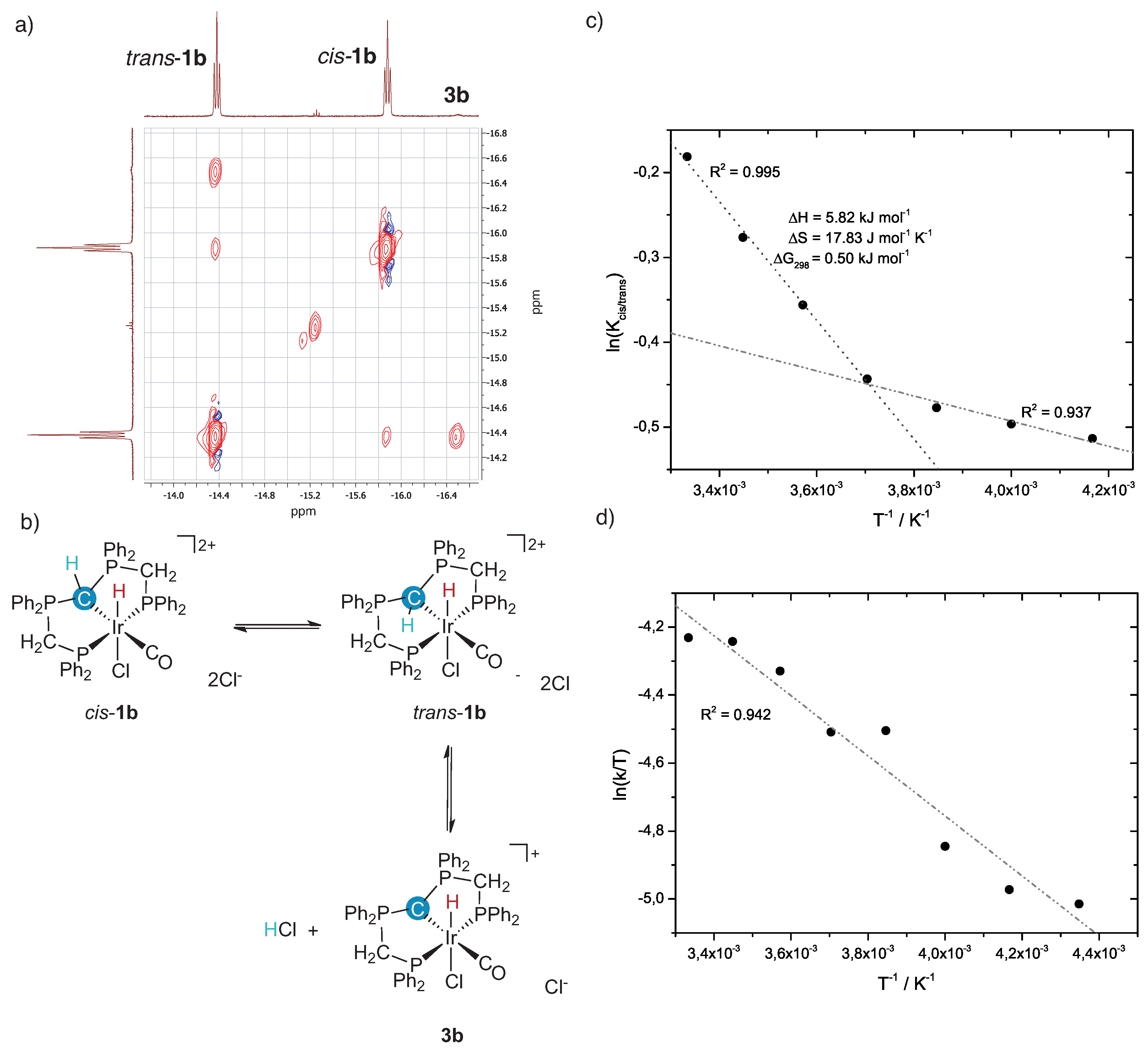
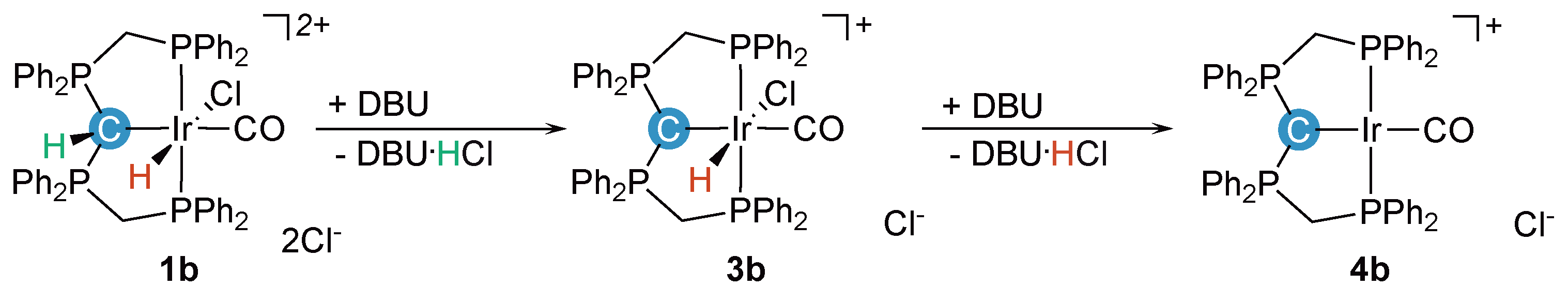
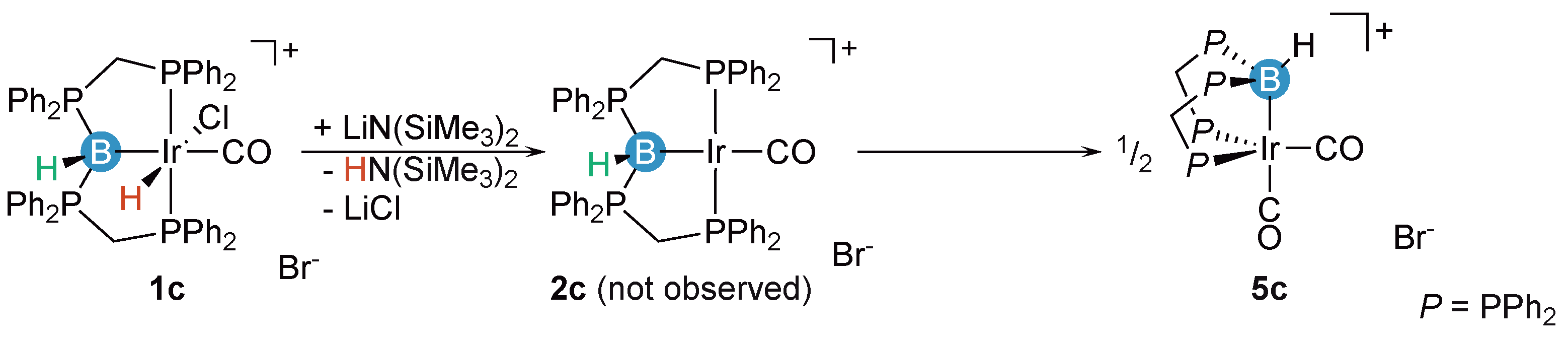
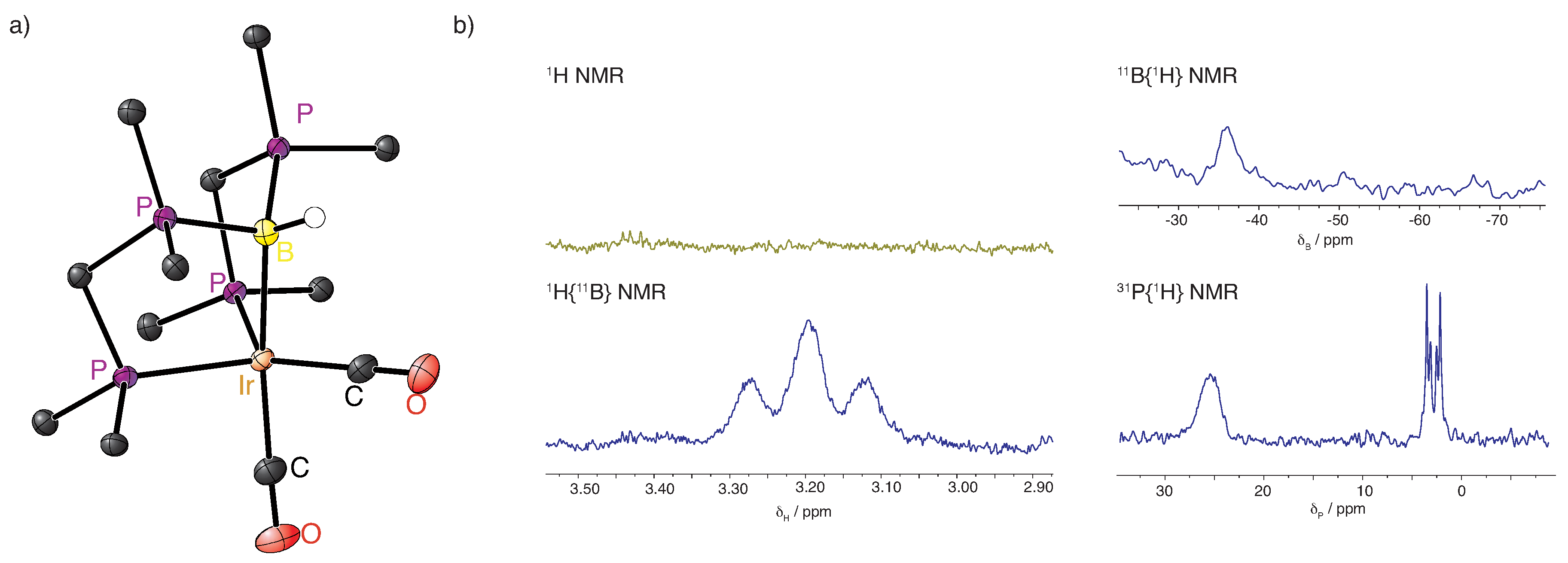
2.1. Deprotonation vs. Reductive Elimination
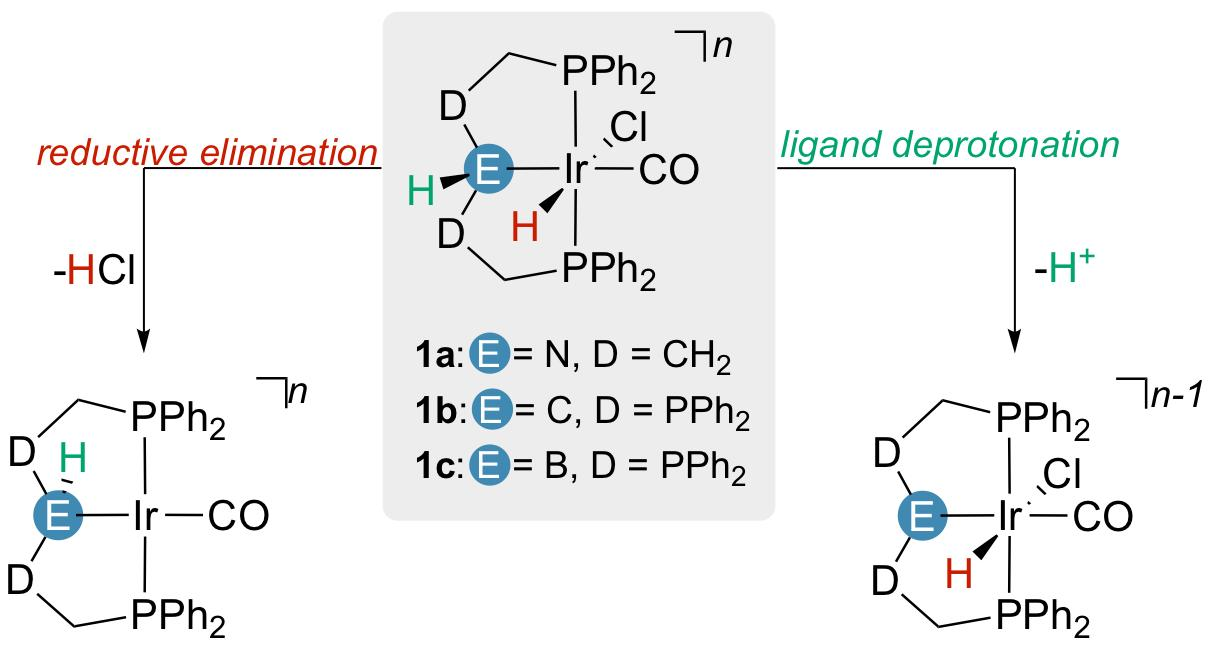
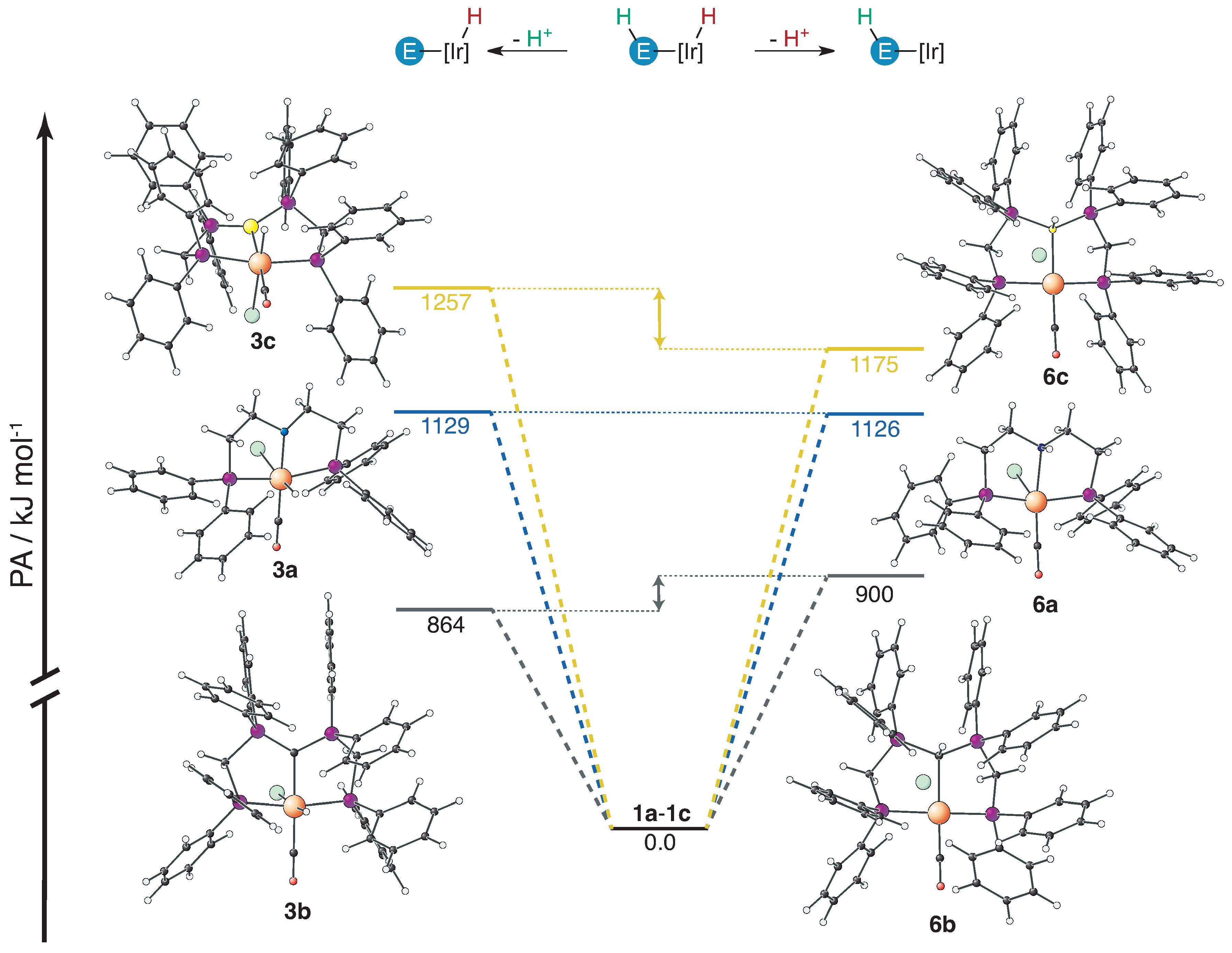
2.2. Proton Affinities and Deprotonation Pathways
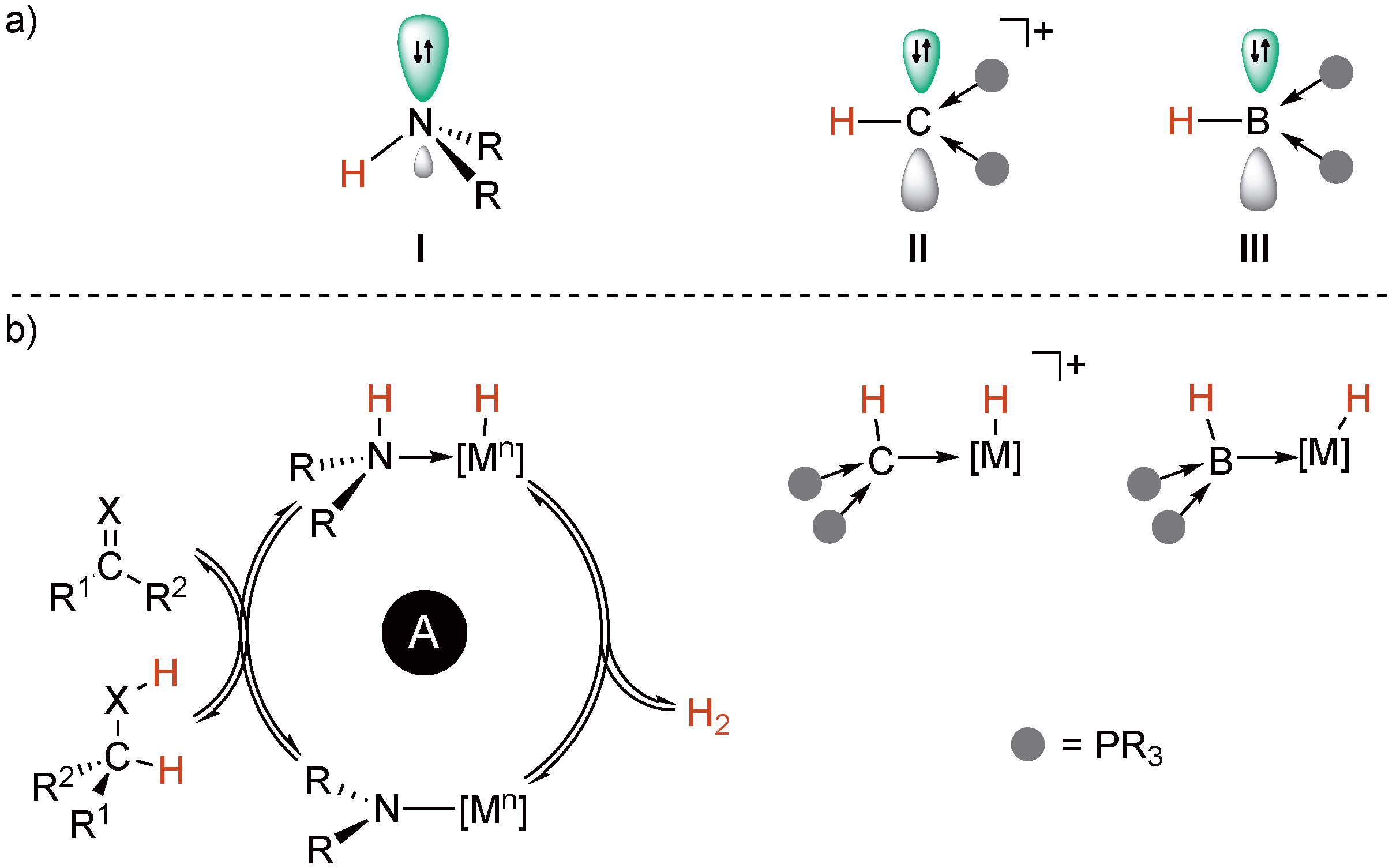
2.3. Comparison with Related Iridium(I) Dicarbonyl Complexes
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDP | carbodiphosphorane |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| dppm | 1,1-bis(diphenylphosphino)methane |
| DFT | density functional theory |
| ESI | electro spray ionisation |
| HMDS | hexamethyldisilazane |
| NMR | nuclear magnetic resonance |
| HRMS | high resolution mass spectrometry |
| THF | tetrahydrofurane |
References
- Bouhadir, G.; Bourissou, D. Complexes of ambiphilic ligands: reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. [Google Scholar] [CrossRef]
- Amgoune, A.; Bourissou, D. σ-Acceptor, Z-type ligands for transition metals. Chem. Commun. 2011, 47, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, H.; Dewhurst, R.D.; Hupp, F.; Nutz, M.; Radacki, K.; Tate, C.W.; Vargas, A.; Ye, Q. Multiple complexation of CO and related ligands to a main-group element. Nature 2015, 522, 327–330. [Google Scholar] [CrossRef]
- Landmann, J.; Sprenger, J.A.P.; Bertermann, R.; Ignat’ev, N.; Bernhardt-Pitchougina, V.; Bernhardt, E.; Willner, H.; Finze, M. Convenient access to the tricyanoborate dianion and selected reactions as a boron-centred nucleophile. Chem. Commun. 2015, 51, 4989–4992. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, E.; Bernhardt-Pitchougina, V.; Willner, H.; Ignatiev, N. “Umpolung” at boron by reduction of [B(CN)4]− and formation of the dianion [B(CN)3]2−. Angew. Chem. Int. Ed. 2011, 50, 12085–12088. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, R.; Donnadieu, B.; Celik, M.A.; Frenking, G.; Bertrand, G. Synthesis and characterization of a neutral tricoordinate organoboron isoelectronic with amines. Science 2011, 333, 610–613. [Google Scholar] [CrossRef]
- Ruiz, D.A.; Melaimi, M.; Bertrand, G. An efficient synthetic route to stable bis(carbene)borylenes [(L1)(L2)BH]. Chem. Commun. 2014, 50, 7837–7839. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Li, Y.; Ganguly, R.; Vidovic, D.; Kinjo, R. Isolation of a Bis(oxazol-2-ylidene)-phenylborylene adduct and its reactivity as a boron-centered nucleophile. Angew. Chem. Int. Ed. 2014, 53, 9280–9283. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Ganguly, R.; Li, Y.; Kinjo, R. Diverse reactivity of a tricoordinate organoboron L2PhB: (L = oxazol-2-ylidene) towards alkali metal, group 9 metal, and coinage metal precursors. Chem. Sci. 2015, 6, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, D.A.; Ung, G.; Melaimi, M.; Bertrand, G. Deprotonation of a Borohydride: Synthesis of a Carbene-Stabilized Boryl Anion. Angew. Chem. Int. Ed. 2013, 52, 7590–7592. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The mechanism of enantioselective ketone reduction with Noyori and Noyori–Ikariya bifunctional catalysts. Dalton Trans. 2016, 45, 6756–6781. [Google Scholar] [CrossRef]
- Maser, L.; Herritsch, J.; Langer, R. Carbodiphosphorane-based nickel pincer complexes and their (de)protonated analogues: Dimerisation, ligand tautomers and proton affinities. Dalton Trans. 2018, 47, 10544–10552. [Google Scholar] [CrossRef] [PubMed]
- Vondung, L.; Frank, N.; Fritz, M.; Alig, L.; Langer, R. Phosphine-Stabilized Borylenes and Boryl Anions as Ligands? Redox Reactivity in Boron-Based Pincer Complexes. Angew. Chem. Int. Ed. 2016, 55, 14450–14454. [Google Scholar] [CrossRef] [PubMed]
- Vondung, L.; Jerabek, P.; Langer, R. Ligands Based on Phosphine-Stabilized Aluminum(I), Boron(I), and Carbon(0). Chem. Eur. J. 2019, 25, 3068–3076. [Google Scholar] [CrossRef] [PubMed]
- Landmann, J.; Keppner, F.; Hofmann, D.B.; Sprenger, J.A.; Höring, M.; Zottnick, S.H.; Müller-Buschbaum, K.; Ignat’ev, N.V.; Finze, M. Deprotonation of a Hydridoborate Anion. Angew. Chem. Int. Ed. 2017, 56, 2795–2799. [Google Scholar] [CrossRef]
- Maser, L.; Schneider, C.; Vondung, L.; Alig, L.; Langer, R. Quantifying the Donor Strength of Ligand-Stabilized Main Group Fragments. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Friedrich, A.; Ghosh, R.; Kolb, R.; Herdtweck, E.; Schneider, S. Iridium olefin complexes bearing dialkylamino/amido PNP pincer ligands: Synthesis, reactivity, and solution dynamics. Organometallics 2009, 28, 708–718. [Google Scholar] [CrossRef]
- Schneider, S.; Meiners, J.; Askevold, B. Cooperative aliphatic PNP amido pincer ligands-versatile building blocks for coordination chemistry and catalysis. Eur. J. Inorg. Chem. 2012, 2012, 412–429. [Google Scholar] [CrossRef]
- Cheng, F.; Yang, X.; Peng, H.; Chen, D.; Jiang, M. Well-controlled formation of polymeric micelles with a nanosized aqueous core and their applications as nanoreactors. Macromolecules 2007, 40, 8007–8014. [Google Scholar] [CrossRef]
- Morris, R.H. Estimating the acidity of transition metal hydride and dihydrogen complexes by adding ligand acidity constants. J. Am. Chem. Soc. 2014, 136, 1948–1959. [Google Scholar] [CrossRef]
- Grätz, M.; Bäcker, A.; Vondung, L.; Maser, L.; Reincke, A.; Langer, R. Donor ligands based on tricoordinate boron formed by B–H-activation of bis(phosphine)boronium salts. Chem. Commun. 2017, 53, 7230–7233. [Google Scholar] [CrossRef] [PubMed]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C.; Trans, D.; Addison, A.W.; Rao, T.N. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6- dithiaheptane]copper(II) perchlorate. Dalton Trans. J. Chem. Soc. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Heins, W.; Mayer, H.A.; Fawzi, R.; Steimann, M. Stereochemical and Electronic Control of Functionalized Tripodal Phosphines. Reactivity of the Adamantane-Type Ir(tripod)(CO)Cl (tripod = cis,cis-1,3,5-(PPh2)3-1,3,5-X3C6H6; X = H, COOMe, CN) Complexes toward H+, H2, CO, and C2H4. Organometallics 1996, 15, 3393–3403. [Google Scholar] [CrossRef]
- Fox, D.J.; Duckett, S.B.; Flaschenriem, C.; Brennessel, W.W.; Schneider, J.; Gunay, A.; Eisenberg, R. A Model Iridium Hydroformylation System with the Large Bite Angle Ligand Xantphos: Reactivity with Parahydrogen and Implications for Hydroformylation Catalysis. Inorg. Chem. 2006, 45, 7197–7209. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, V.; Pérez-Torrente, J.J.; Oro, L.A. Intramolecular C–H Oxidative Addition to Iridium(I) in Complexes Containing a N,N′-Diphosphanosilanediamine Ligand. Inorg. Chem. 2014, 53, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Chianese, A.R.; Li, X.; Janzen, M.C.; Faller, J.W.; Crabtree, R.H. Rhodium and iridium complexes of N-heterocyclic carbenes via transmetalation: Structure and dynamics. Organometallics 2003, 22, 1663–1667. [Google Scholar] [CrossRef]
- Kelly III, R.A.; Clavier, H.; Giudice, S.; Scott, N.M.; Stevens, E.D.; Bordner, J.; Samardjiev, I.; Hoff, C.D.; Cavallo, L.; Nolan, S.P. Determination of N-Heterocyclic Carbene (NHC) Steric and Electronic Parameters using the [(NHC)Ir(CO)2Cl] System. Organometallics 2008, 27, 202–210. [Google Scholar] [CrossRef]
- Wolf, S.; Plenio, H. Synthesis of (NHC)Rh(cod)Cl and (NHC)RhCl(CO)2 complexes—Translation of the Rh- into the Ir-scale for the electronic properties of NHC ligands. J. Organomet. Chem. 2009, 694, 1487–1492. [Google Scholar] [CrossRef]









| Donor in 1 | Reactivity | PA(3)/kJ·mol−1 | Reactivity | PA(6)/kJ·mol−1 | ΔPA/kJ·mol−1 |
|---|---|---|---|---|---|
| R2NH | 1a→3a | 1129 | 1a→6a | 1126 | 3 |
| (Ph2RP)2CH | 1b→3b | 864 | 1b→6b | 900 | −36 |
| (Ph2RP)2BH | 1c→3c | 1257 | 1c→6c | 1175 | 82 |





{kind=link}
{kind=link}
{kind=link}
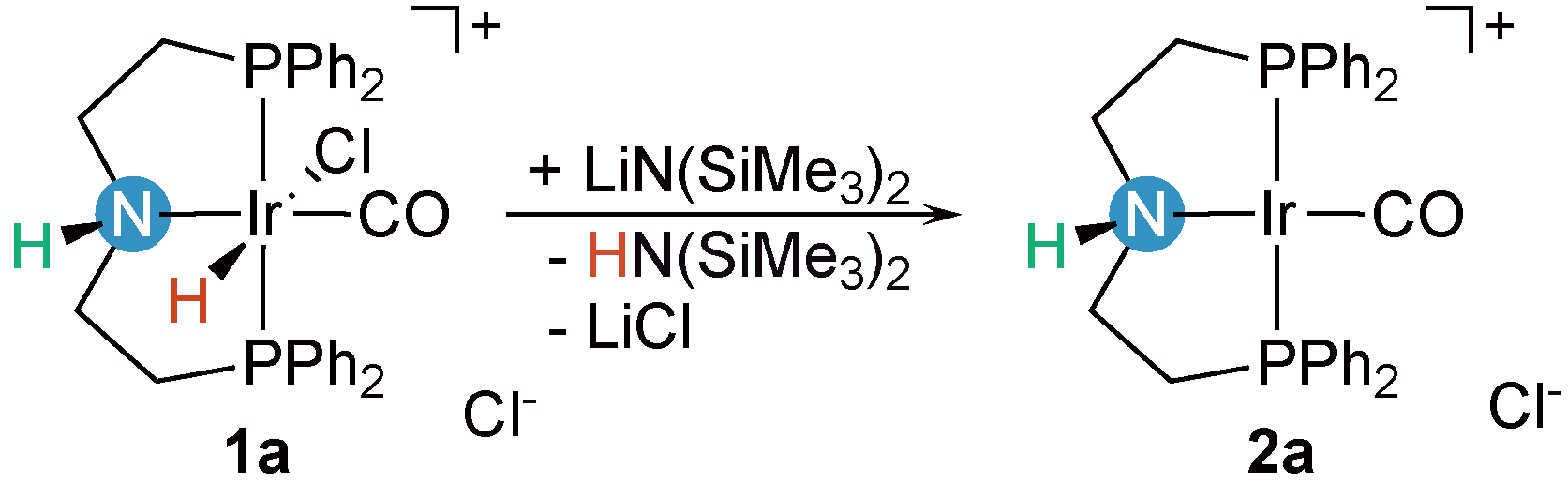{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maser, L.; Schneider, C.; Alig, L.; Langer, R. Comparing the Acidity of (R3P)2BH-Based Donor Groups in Iridium Pincer Complexes. Inorganics 2019, 7, 61. https://doi.org/10.3390/inorganics7050061
Maser L, Schneider C, Alig L, Langer R. Comparing the Acidity of (R3P)2BH-Based Donor Groups in Iridium Pincer Complexes. Inorganics. 2019; 7(5):61. https://doi.org/10.3390/inorganics7050061
Chicago/Turabian StyleMaser, Leon, Christian Schneider, Lukas Alig, and Robert Langer. 2019. "Comparing the Acidity of (R3P)2BH-Based Donor Groups in Iridium Pincer Complexes" Inorganics 7, no. 5: 61. https://doi.org/10.3390/inorganics7050061
APA StyleMaser, L., Schneider, C., Alig, L., & Langer, R. (2019). Comparing the Acidity of (R3P)2BH-Based Donor Groups in Iridium Pincer Complexes. Inorganics, 7(5), 61. https://doi.org/10.3390/inorganics7050061