Molecular Modelling of the Ni(II)-Responsive Synechocystis PCC 6803 Transcriptional Regulator InrS in the Metal Bound Form



Abstract
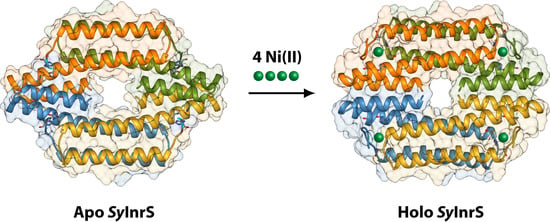
:
1. Introduction
2. Results and Discussion
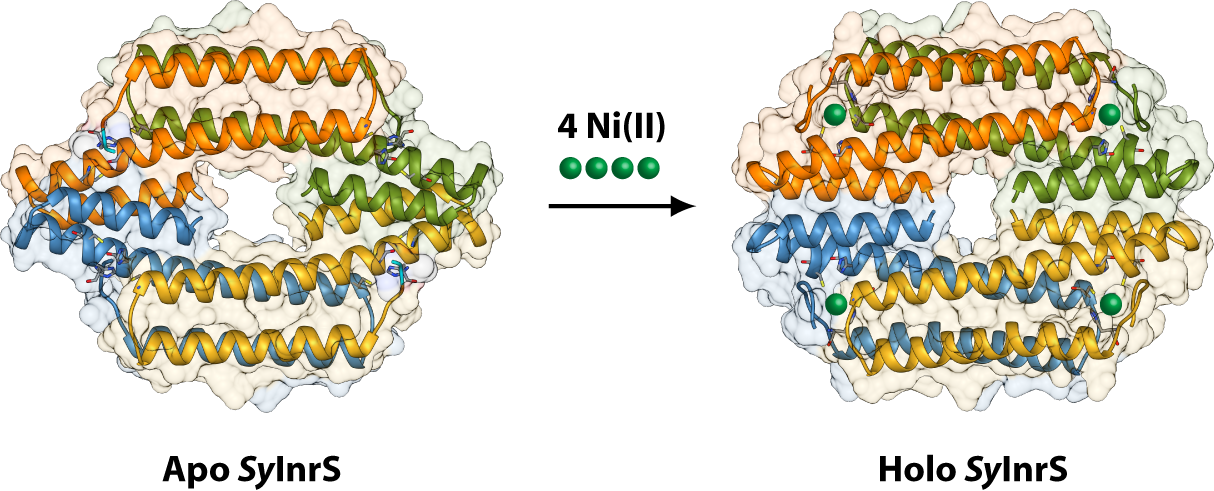
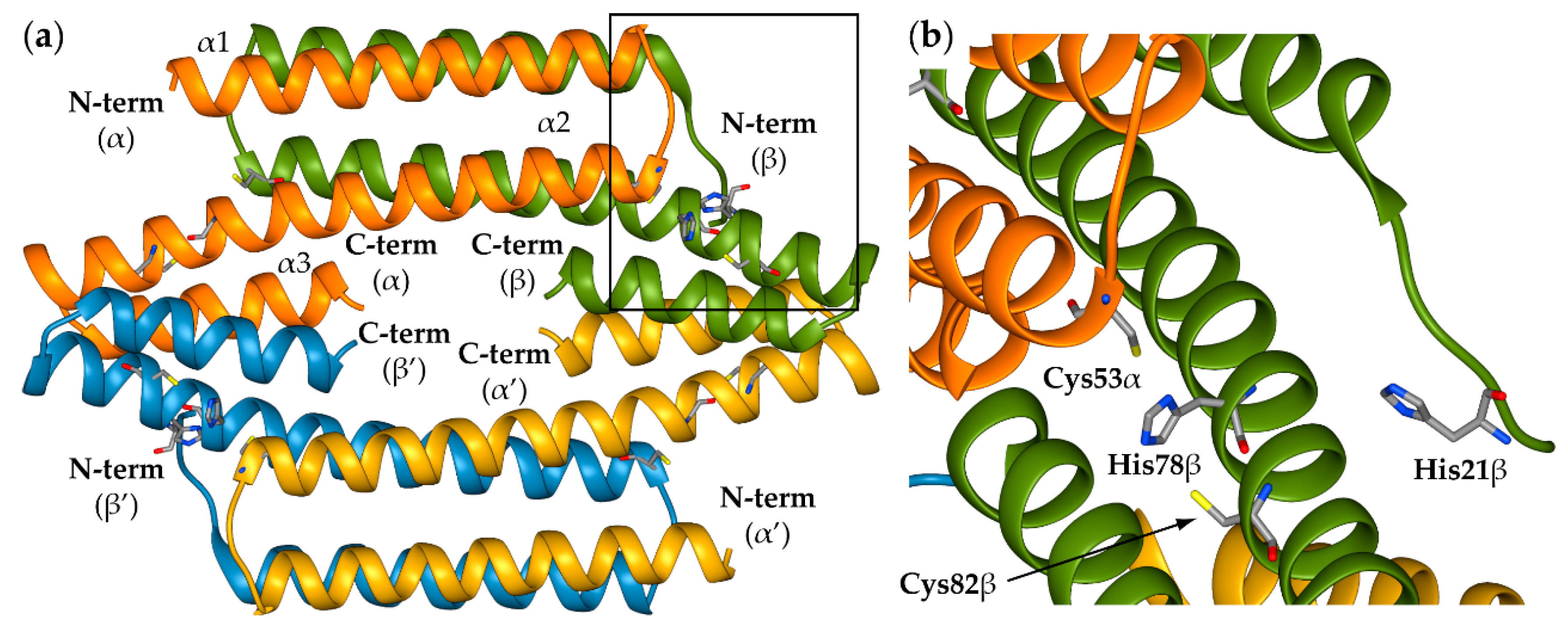
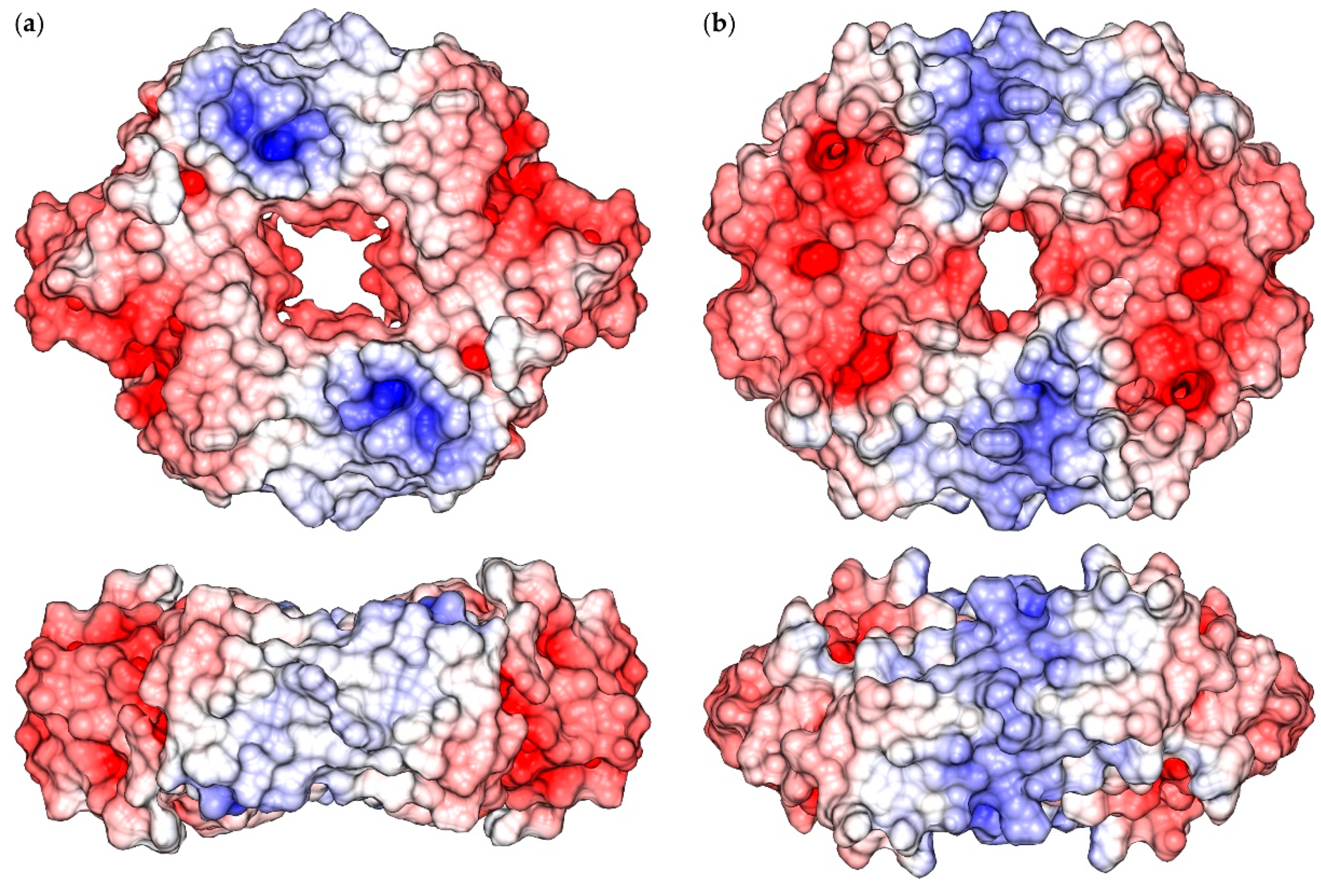
2.1. Reconstruction of Apo-SyInrS N-terminal Region and Modelling of Holo-SyInrS
2.2. Modelling of Ni(II) Bound Forms of SyInrS
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NTA: Nitrilotriacetic acid (2,2′,2′′-Nitrilotriacetic acid) |
| EGTA: Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid |
| EDTA: Ethylenediaminetetraacetic acid (2,2′,2″,2‴-(Ethane-1,2-diyldinitrilo)tetraacetic acid) |
References
- Zamble, D.; Rowińska-Żyrek, M.; Kozlowski, H. The Biological Chemistry of Nickel; The Royal Society of Chemistry: London, UK, 2017; Volume 10, pp. 1–380. [Google Scholar]
- Organization, W.H. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: https://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/ (accessed on 27 January 2017).
- Huertas, M.J.; Lopez-Maury, L.; Giner-Lamia, J.; Sanchez-Riego, A.M.; Florencio, F.J. Metals in cyanobacteria: Analysis of the copper, nickel, cobalt and arsenic homeostasis mechanisms. Life (Basel) 2014, 4, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.W.; Patterson, C.J.; Pernil, R.; Hess, C.R.; Robinson, N.J. Cytosolic Ni(II) sensor in cyanobacterium: Nickel detection follows nickel affinity across four families of metal sensors. J. Biol. Chem. 2012, 287, 12142–12151. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, B.; Musiani, F.; Ciurli, S. Metal ion-mediated DNA-protein interactions. Met. Ions Life Sci. 2012, 10, 135–170. [Google Scholar] [PubMed]
- Musiani, F.; Zambelli, B.; Bazzani, M.; Mazzei, L.; Ciurli, S. Nickel-responsive transcriptional regulators. Metallom. Integr. Biometal Sci. 2015, 7, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ramesh, A.; Ma, Z.; Ward, S.K.; Zhang, L.; George, G.N.; Talaat, A.M.; Sacchettini, J.C.; Giedroc, D.P. CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat. Chem. Biol. 2007, 3, 60–68. [Google Scholar] [CrossRef]
- Chang, F.M.; Coyne, H.J.; Cubillas, C.; Vinuesa, P.; Fang, X.; Ma, Z.; Ma, D.; Helmann, J.D.; Garcia-de los Santos, A.; Wang, Y.X.; et al. Cu(I)-mediated allosteric switching in a copper-sensing operon repressor (CsoR). J. Biol. Chem. 2014, 289, 19204–19217. [Google Scholar] [CrossRef]
- Higgins, K.A.; Giedroc, D. Insights into protein allostery in the CsoR/RcnR family of transcriptional repressors. Chem. Lett. 2014, 43, 20–25. [Google Scholar] [CrossRef]
- Capdevila, D.A.; Edmonds, K.A.; Giedroc, D.P. Metallochaperones and metalloregulation in bacteria. Essays Biochem. 2017, 61, 177–200. [Google Scholar] [CrossRef]
- Herring, C.D.; Blattner, F.R. Global transcriptional effects of a suppressor tRNA and the inactivation of the regulator frmR. J. Bacteriol. 2004, 186, 6714–6720. [Google Scholar] [CrossRef]
- Denby, K.J.; Iwig, J.; Bisson, C.; Westwood, J.; Rolfe, M.D.; Sedelnikova, S.E.; Higgins, K.; Maroney, M.J.; Baker, P.J.; Chivers, P.T.; et al. The mechanism of a formaldehyde-sensing transcriptional regulator. Sci. Rep. 2016, 6, 38879. [Google Scholar] [CrossRef] [Green Version]
- Grossoehme, N.; Kehl-Fie, T.E.; Ma, Z.; Adams, K.W.; Cowart, D.M.; Scott, R.A.; Skaar, E.P.; Giedroc, D.P. Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J. Biol. Chem. 2011, 286, 13522–13531. [Google Scholar] [CrossRef]
- Luebke, J.L.; Shen, J.; Bruce, K.E.; Kehl-Fie, T.E.; Peng, H.; Skaar, E.P.; Giedroc, D.P. The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol. 2014, 94, 1343–1360. [Google Scholar] [CrossRef] [PubMed]
- Chivers, P.T. Nickel Regulation. In The Biological Chemistry of Nickel; The Royal Society of Chemistry: London, UK, 2017; pp. 259–283. [Google Scholar]
- Foster, A.W.; Pernil, R.; Patterson, C.J.; Robinson, N.J. Metal specificity of cyanobacterial nickel-responsive repressor InrS: Cells maintain zinc and copper below the detection threshold for InrS. Mol. Microbiol. 2014, 92, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.W.; Pernil, R.; Patterson, C.J.; Scott, A.J.P.; Pålsson, L.-O.; Pal, R.; Cummins, I.; Chivers, P.T.; Pohl, E.; Robinson, N.J. A tight tunable range for Ni(II) sensing and buffering in cells. Nat. Chem. Biol. 2017, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.E.; Foster, A.W.; Maroney, M.J. An XAS investigation of the nickel site structure in the transcriptional regulator InrS. J. Inorg. Biochem. 2017, 177, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucl. Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Tan, B.G.; Vijgenboom, E.; Worrall, J.A. Conformational and thermodynamic hallmarks of DNA operator site specificity in the copper sensitive operon repressor from Streptomyces lividans. Nucl. Acids Res. 2014, 42, 1326–1340. [Google Scholar] [CrossRef]
- Chang, F.M.; Lauber, M.A.; Running, W.E.; Reilly, J.P.; Giedroc, D.P. Ratiometric pulse-chase amidination mass spectrometry as a probe of biomolecular complex formation. Anal. Chem. 2011, 83, 9092–9099. [Google Scholar] [CrossRef]
- Porto, T.V.; Hough, M.A.; Worrall, J.A. Structural insights into conformational switching in the copper metalloregulator CsoR from Streptomyces lividans. Acta Crystallogr. D 2015, 71, 1872–1878. [Google Scholar] [CrossRef]
- Chang, F.M.; Martin, J.E.; Giedroc, D.P. Electrostatic occlusion and quaternary structural ion pairing are key determinants of Cu(I)-mediated allostery in the copper-sensing operon repressor (CsoR). Biochemistry 2015, 54, 2463–2472. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Hutchinson, E.G.; Michie, A.D.; Wallace, A.C.; Jones, M.L.; Thornton, J.M. PDBsum: A web-based database of summaries and analyses of all PDB structures. Trends. Biochem. Sci. 1997, 22, 488–490. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.E.; Musiani, F.; Huang, H.T.; Chivers, P.T.; Ciurli, S.; Maroney, M.J. Glutamate ligation in the Ni(II)- and Co(II)-responsive Escherichia coli transcriptional regulator, RcnR. Inorg. Chem. 2017, 56, 6459–6476. [Google Scholar] [CrossRef]
- Martin-Diaconescu, V.; Bellucci, M.; Musiani, F.; Ciurli, S.; Maroney, M.J. Unraveling the Helicobacter pylori UreG zinc binding site using X-ray absorption spectroscopy (XAS) and structural modeling. J. Biol. Inorg. Chem. 2012, 17, 353–361. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.T.; Bobst, C.E.; Iwig, J.S.; Chivers, P.T.; Kaltashov, I.A.; Maroney, M.J. Co(II) and Ni(II) binding of the Escherichia coli transcriptional repressor RcnR orders its N terminus, alters helix dynamics, and reduces DNA affinity. J. Biol. Chem. 2018, 293, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Sala, D.; Musiani, F.; Rosato, A. Application of molecular dynamics to the investigation of metalloproteins involved in metal homeostasis. Eur. J. Inorg. Chem. 2018, 2018, 4661–4677. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Miyashita, N.; Im, W.; Feig, M.; Sugita, Y. Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Biophys. Acta 2016, 1858, 1635–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, M. A theoretical view of protein dynamics. Chem. Soc. Rev. 2014, 43, 5051–5066. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: Several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.A.; Huang, Y.M.; Mueller, L.J.; You, W. Investigation of structural dynamics of enzymes and protonation states of substrates using computational tools. Catalysts 2016, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Musiani, F.; Bernetti, M.; Falchi, F.; Cavalli, A.; Ciurli, S.; Recanatini, M. Development of a multisite model for Ni(II) ion in solution from thermodynamic and kinetic data. J. Comput. Chem. 2017, 38, 1834–1843. [Google Scholar] [CrossRef]
- Van Dijk, M.; van Dijk, A.D.; Hsu, V.; Boelens, R.; Bonvin, A.M. Information-driven protein-DNA docking using HADDOCK: It is a matter of flexibility. Nucl. Acids Res. 2006, 34, 3317–3325. [Google Scholar] [CrossRef]
- Van Dijk, M.; Bonvin, A.M. Pushing the limits of what is achievable in protein-DNA docking: Benchmarking HADDOCK’s performance. Nucl. Acids Res. 2010, 38, 5634–6547. [Google Scholar] [CrossRef] [PubMed]
- Musiani, F.; Ciurli, S. Evolution of macromolecular docking techniques: The case study of nickel and iron metabolism in pathogenic bacteria. Molecules 2015, 20, 14265–14292. [Google Scholar] [CrossRef] [PubMed]
- Agriesti, F.; Roncarati, D.; Musiani, F.; Del Campo, C.; Iurlaro, M.; Sparla, F.; Ciurli, S.; Danielli, A.; Scarlato, V. FeON-FeOFF: The Helicobacter pylori Fur regulator commutates iron-responsive transcription by discriminative readout of opposed DNA grooves. Nucl. Acids Res. 2014, 42, 3138–3151. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, L.; Dobrovolska, O.; Musiani, F.; Zambelli, B.; Ciurli, S. On the interaction of Helicobacter pylori NikR, a Ni(II)-responsive transcription factor, with the urease operator: in solution and in silico studies. J. Biol. Inorg. Chem. 2015, 20, 1021–1037. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformation | Interacting Monomers | Interface Residues | Interface Surface (Å2) | Non-Bonded Contacts | Hydrogen Bonds | Salt Bridges |
|---|---|---|---|---|---|---|
| Apo | α-β (α’-β’) | 22 | 1490 | 87 | 8 | 6 |
| α-β’ (α’-β) | 17 | 952 | 55 | 5 | 2 | |
| Holo | α-β (α’-β’) | 30 | 1950 | 133 | 2 | 4 |
| α-β’ (α’-β) | 13 | 670 | 78 | 0 | 2 |
| Constrained Atoms | Distance (Å) | |
| Ni(II)–His21(Nδ/ε) | 1.9 ± 0.05 | |
| Ni(II)–His78(Nδ/ε) | 1.9 ± 0.05 | |
| Ni(II)–Cys53(Sγ) | 2.2 ± 0.05 | |
| Ni(II)–His82(Sγ) | 2.2 ± 0.05 | |
| Bonded Atoms | Constrained Atoms | Angle (Degrees) |
| Ni(II)–Cys(Sγ) | Ni(II)–Cys(Sγ)–Cys(Cβ) | 109 ± 5 |
| Ni(II)–His(Nδ) | Ni(II)–His(Nδ)–His(Cγ) | 120 ± 10 |
| Ni(II)–His(Nδ)–His(Cε) | 120 ± 10 | |
| Ni(II)–His(Nε) | Ni(II)–His(Nε)–His(Cδ) | 120 ± 10 |
| Ni(II)–His(Nε)–His(Cε) | 120 ± 10 | |
| Bonded Atoms | Constrained Atoms | Dihedral (Degrees) |
| Ni(II)–His(Nδ) | Ni(II)–His(Nδ)–His(Cε)–His(Nε) | 180 ± 10 |
| Ni(II)–His(Nδ)–His(Cγ)–His(Cδ) | 180 ± 10 | |
| Ni(II)–His(Nε) | Ni(II)–His(Nε)–His(Cε)–His(Nδ) | 180 ± 10 |
| Ni(II)−His(Nε)–His(Cδ)–His(Cγ) | 180 ± 10 | |
| Initial Conformation | Apo-SyInrS | Holo-SyInrS | ||
|---|---|---|---|---|
| Model | Best Coordination Geometry | RMSD (Å) | Best Conformation Geometry | RMSD (Å) |
| H21(Nε)–H78(Nδ) | Tetrahedral | 0.54 | Square planar | 0.29 |
| H21(Nδ)–H78(Nε) | Tetrahedral | 0.23 | Tetrahedral | 0.53 |
| H21(Nδ)–H78(Nδ) | Tetrahedral | 0.65 | Tetrahedral | 0.36 |
| H21(Nε)–H78(Nε) | Tetrahedral | 0.39 | Square planar | 0.27 |
| Ramachandran Plot Region | Holo-SyInrS Ni(II) Bound Model | |
|---|---|---|
| H21(Nδ)–H78(Nε) | H21(Nε)–H78(Nε) | |
| Most favoured | 94.2% | 90.7% |
| Additionally allowed | 5.8% | 9.3% |
| Generously allowed | - | - |
| Disallowed | - | - |
| G-factor | 0.09 | 0.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barchi, E.; Musiani, F. Molecular Modelling of the Ni(II)-Responsive Synechocystis PCC 6803 Transcriptional Regulator InrS in the Metal Bound Form. Inorganics 2019, 7, 76. https://doi.org/10.3390/inorganics7060076
Barchi E, Musiani F. Molecular Modelling of the Ni(II)-Responsive Synechocystis PCC 6803 Transcriptional Regulator InrS in the Metal Bound Form. Inorganics. 2019; 7(6):76. https://doi.org/10.3390/inorganics7060076
Chicago/Turabian StyleBarchi, Elia, and Francesco Musiani. 2019. "Molecular Modelling of the Ni(II)-Responsive Synechocystis PCC 6803 Transcriptional Regulator InrS in the Metal Bound Form" Inorganics 7, no. 6: 76. https://doi.org/10.3390/inorganics7060076
APA StyleBarchi, E., & Musiani, F. (2019). Molecular Modelling of the Ni(II)-Responsive Synechocystis PCC 6803 Transcriptional Regulator InrS in the Metal Bound Form. Inorganics, 7(6), 76. https://doi.org/10.3390/inorganics7060076