1. Introduction
Fossil fuels play a dominant role in chemical sciences and industries. However, fossil fuel dependency is unsustainable due to its limited amount of resources. Moreover, a large number of greenhouse gases, such as CO
2, are emitted by the use of fossil fuels. Biomass has the potential to provide a sustainable and carbon-neutral process which complements the drawback of fossil fuels and possibly suppresses global warming [
1]. In recent years, a number of research directions focusing on biomass transformation have been explored, such as for biofuels and bioplastics [
2]. On the other hand, a trivalent iridium complex bearing a pentamethylcyclopentadienyl (Cp*) ligand is a well-known stable complex [
3]. The Cp* ligand is not susceptible to chemical transformation and has been used as an effective ligand because it can form stable coordination with a metal center in a tridentate coordination fashion. Thus, the Cp* ligand has been employed to improve the stabilities and hemilabilities of catalytically active species [
4]. Therefore, a number of IrCp*-catalyzed reactions have been reported so far [
5]; however, there has been no report on the reduction of carbonyl compounds using biogenetic alcohols as hydrogen sources despite the known reducing property of glucose in the field of metal nanoparticles synthesis [
6]. Though RhCp*-catalyzed hydrogenation using sugars as hydrogen sources has been reported, excess amounts of sugar, bases, and toxic chlorinated solvents were necessary [
7]. Herein, [IrCp*Cl
2]
2-catalyzed reduction of carbonyl groups is reported, which consists of biogenetic alcohols, such as monosaccharides, oligosaccharides, or glycerol, as a hydrogen source. This system does not require any base, and the reaction can proceed in the presence of water.
2. Results and Discussions
The investigation on the optimization of [IrCp*Cl
2]
2-catalyzed reaction condition using 2-naphthaldehyde (
1a) as a substrate was performed. Reductions of
1a using sugars as hydrogen sources under base-free conditions were conducted. The reaction was carried out using different types of solvents in the presence of [IrCp*Cl
2]
2 as a catalyst (5.0 mol %-Ir) and glucose as a hydrogen source at 85 °C for 24 h; as a result, 2-naphthyl methanol (
2a) was formed (
Table 1). In order to dissolve glucose, water was required for this reaction. The choice of solvent was essential for the high conversion of
1a; a less polar solvent such as CH
2Cl
2 and toluene showed lower yields of 45% (Entry 1) and 61% (Entry 2), while a polar solvent such as tetrahydrofuran (THF), MeOH, and 1,4-dioxane gave higher yields of 81% (Entry 3), 93% (Entry 4) and 95% (Entry 5), respectively. When the amount of the catalyst was reduced to 1 mol%, the product yield decreased to 53% (Entry 6). The addition of a base may have had some effect; the product yield slightly increased to 60% when 1 mol % of Ir and 10 mol % of K
2CO
3 were used (Entry 7).
Encouraged by the above results, we envisaged using other sugars in the IrCp*-catalyzed reactions. The results of the investigation are summarized in
Table 2. The reaction of monosaccharides, such as glucose, galactose, and xylose, resulted in excellent yields (Entries 1–3). When the reaction was performed using a disaccharide such as lactose, sucrose, and maltose, similar yields were obtained (Entries 4–6), while decreasing the amount of disaccharide also caused a decrease of the product yield. This suggests a 1:1 reaction of a sugar and
1a despite of the number of hydroxyl groups (Entry 7). Similarly, trisaccharide, and raffinose also gave
2a in a high yield (Entry 8). However, when cellulose was used, the reaction did not proceed. These results suggest that the choice of sugar is also essential.
Next, the reduction reactions of various aldehydes were investigated (
Table 3). As described above,
1a gave
2a in an 88% isolated yield (Entry 1). Noticeable electronic and steric effects were not observed for aromatic aldehydes bearing the 4-methyl (
1b, Entry 2), 4-cyano (
1c, Entry 3), 4-trifluoromethyl (
1d, Entry 4), 4-bromo (
1e, Entry 5), and 2-bromo groups (
1f, Entry 6). Heteroaromatic aldehydes, such as 4-pyridinecarboxaldehyde (
1g, Entry 7) and 2-thiophenecarboxaldehyde (
1h, Entry 8) were converted to the corresponding alcohols. Furthermore, alkyl aldehyde (
1i, Entry 9) and ketone (
1j, Entry 10) could also be applicable. However, any alkenes and alkynes were out of the scope for the catalytic system.
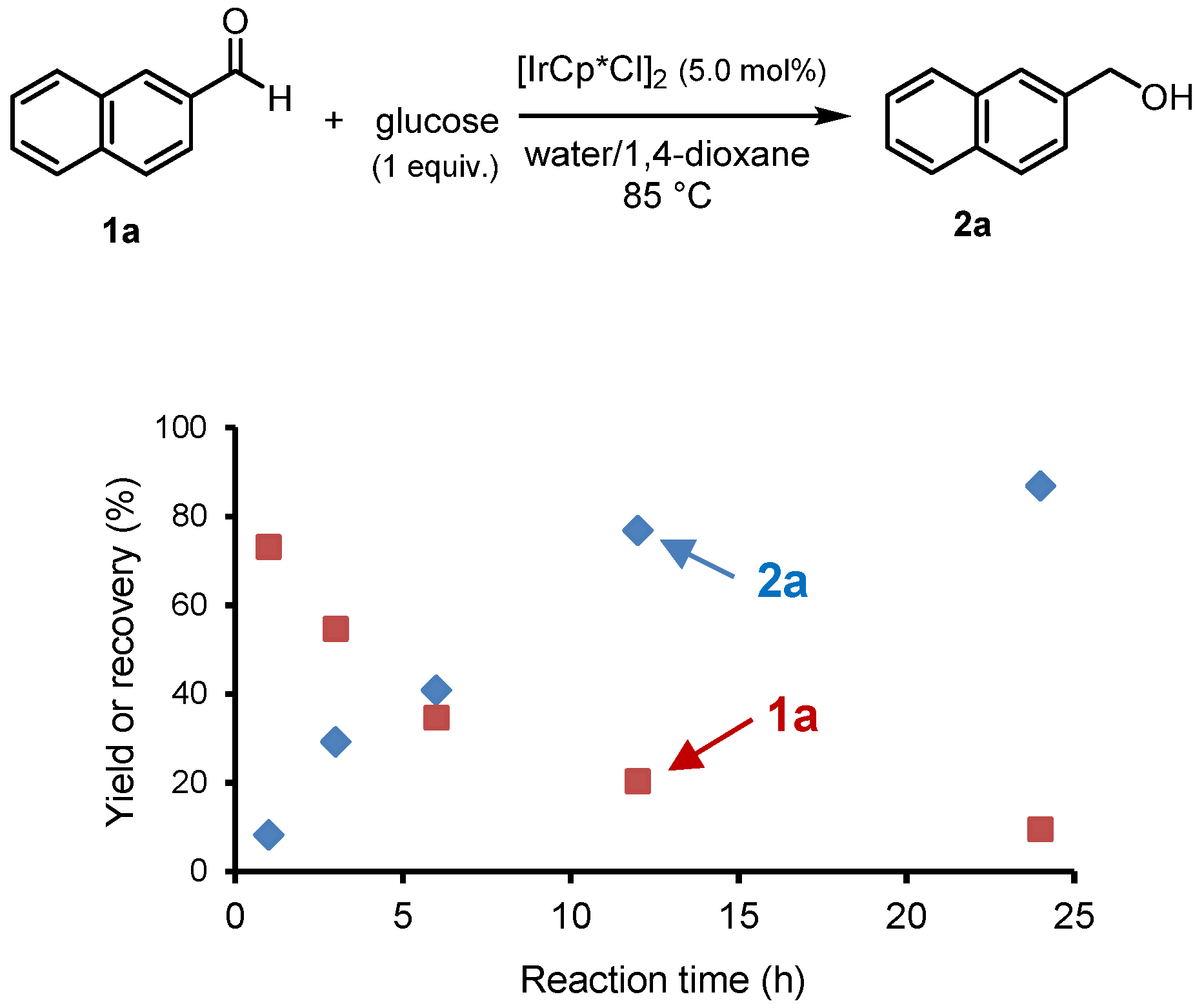
A time-resolved reaction profile for the [IrCp*Cl
2]
2-catalyzed reduction of aldehyde
1a to alcohol
2a is shown in
Figure 1. This profile indicates that the
2a product gradually formed, and a 24 h reaction is necessary.
Next, an investigation was performed to confirm which hydrogen on a sugar was used as a reductant. Using methyl α-
d glucoside, the system did not afford the desired product (
Table 4, Entry 1). However, another biogenetic alcohol, glycerol, worked as a hydrogen source (
Table 4, Entry 2). This suggests that glucose binds to the Ir center via the deprotonated hydroxyl group at the anomeric carbon followed by a hydrogen shift from that carbon to iridium, and then the reduction of the aldehyde may occur.
The reaction mechanism is supposed to be similar to the traditional [IrCp*Cl
2]
2-catalyzed transfer hydrogenation with fossil fuel-derived alcohols [
3]. However, the reason for the lack of need of the base is still unclear. Further investigations would be necessary to distinguish the difference among the reaction mechanisms occurring with different types of alcohol.
3. Materials and Methods
All reactions were carried out under an argon atmosphere. 1,4-dioxane and d(+)-glucose were purchased from Wako Pure Chemical Industries (Osaka, Japan). The Ir complex was purchased from Furuya Metal Co., Ltd. (Tokyo, Japan). Glucono-δ-lactone was purchased from Kishida Chemical Co. (city, country), which was used for the LC–MS analysis of the standard sample. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded using a JEOL JNM-LA400 spectrometer (JEOL, Ltd, Tokyo, Japan). Proton chemical shifts were relative to solvent peaks [chloroform: 7.27 (1H), 77.00 (13C)]. Reactions were monitored by thin-layer chromatography (TLC) that was carried out on 0.25 mm Merck silica gel plates 60F-254 (Merck, Darmstadt, Germany) using UV light for visualization.
The catalytic reaction was performed as follows: Aldehyde (0.25 mmol), sugar (0.25 mmol) and [IrCp*Cl
2]
2 (5.0 mol %) were dissolved in H
2O (0.5 mL) and 1,4-dioxane (0.5 mL). The reaction mixture was stirred for 24 h at 85 °C. After cooling, the reaction mixture was diluted with H
2O and extracted with ethyl acetate. For gas chromatography analysis, a known amount of dodecane was added in the mixture, and the product yield was determined by comparing the areas of the GC spectra. For
1H NMR analysis, the mixture was concentrated under reduced pressure, and the crude
1H-NMR spectra in CDCl
3 was obtained using a known amount of 1,1,2,2-tetrachloroethane as an internal standard (see
Supplementary Materials). The yield was measured by integrating the H of the benzylic position with respect to the 1,1,2,2-tetrachloroethane peak.




{kind=link}
{kind=link}



 1a
1a 1b
1b 1c
1c 1d
1d 1e
1e 1f
1f 1g
1g 1h
1h 1i
1i 1j
1j

