Synthesis, Crystal Structures and Thermal Properties of Ammine Barium Borohydrides




Abstract
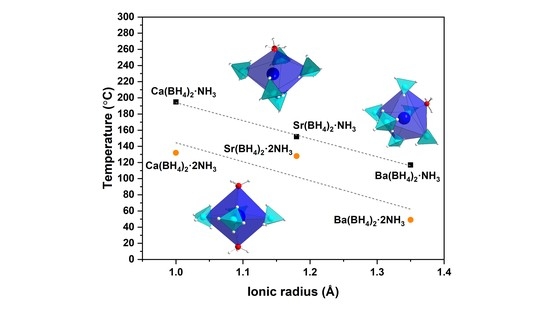
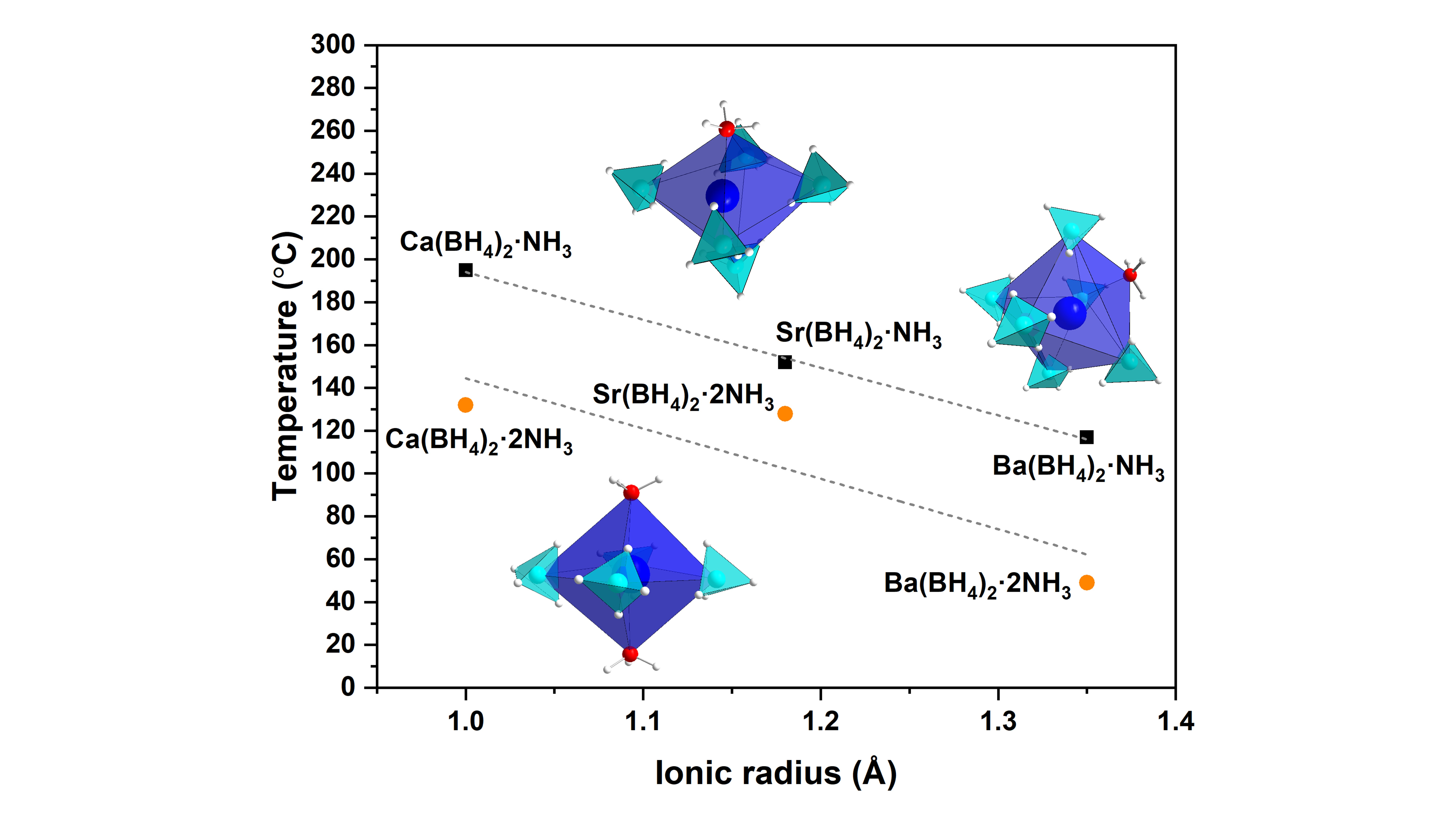
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Initial Phase Analysis
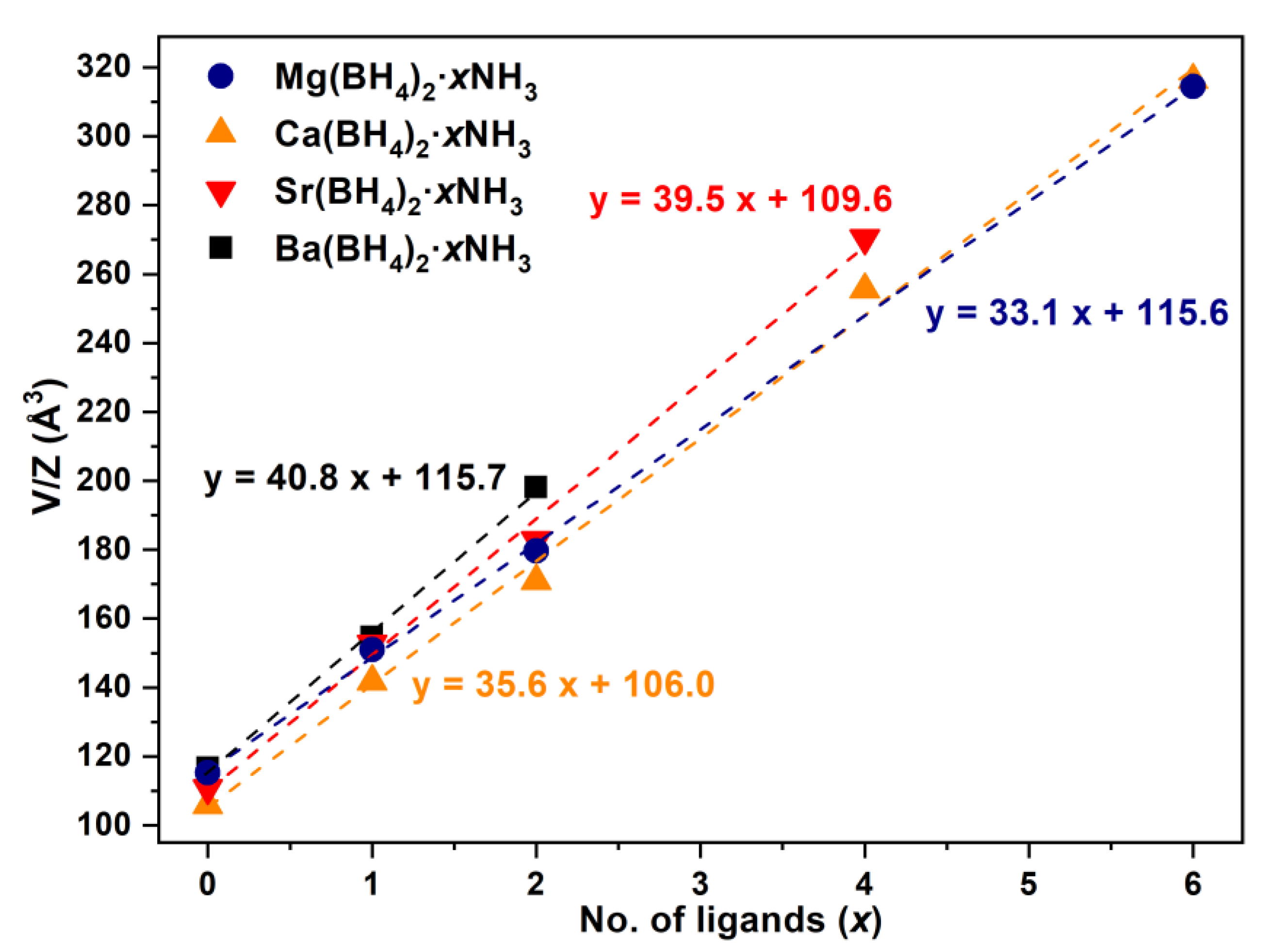
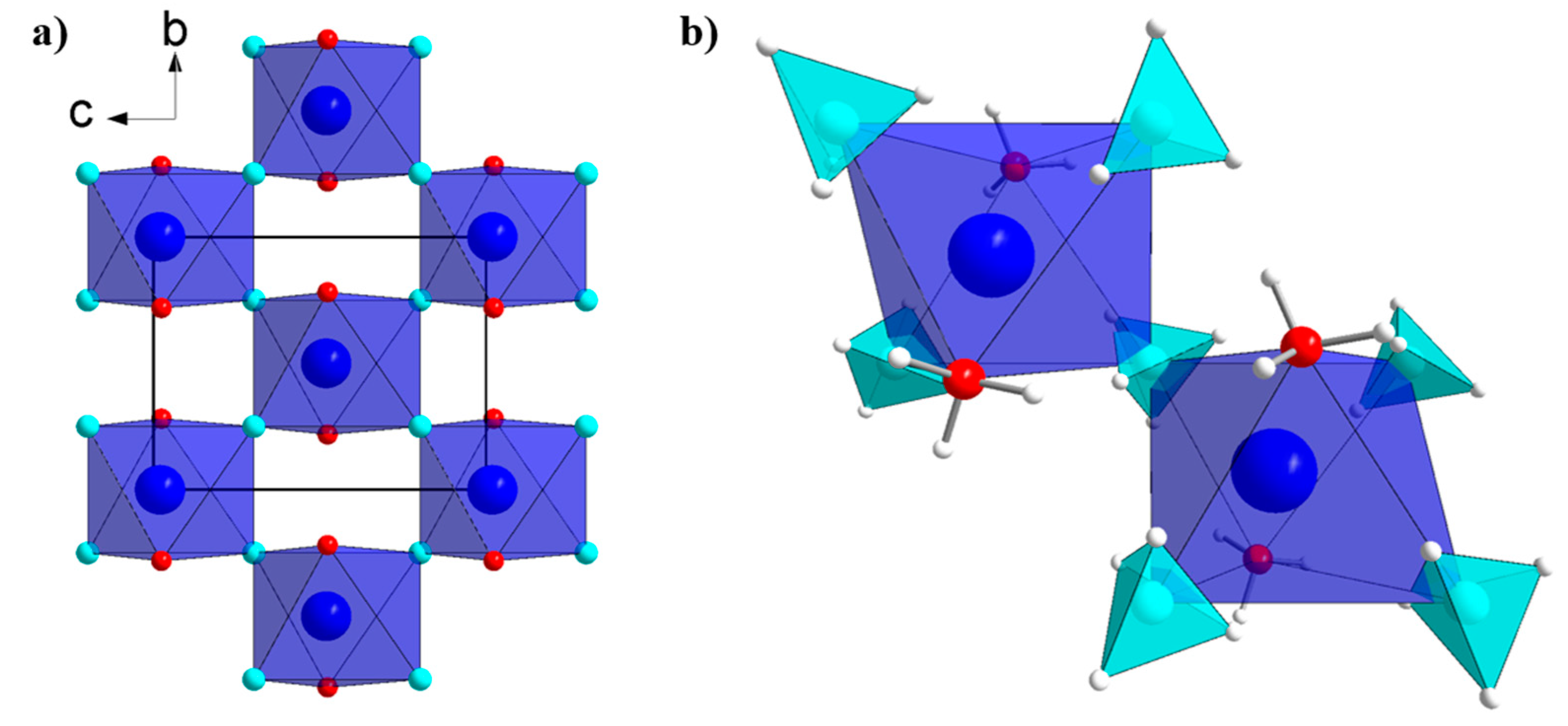
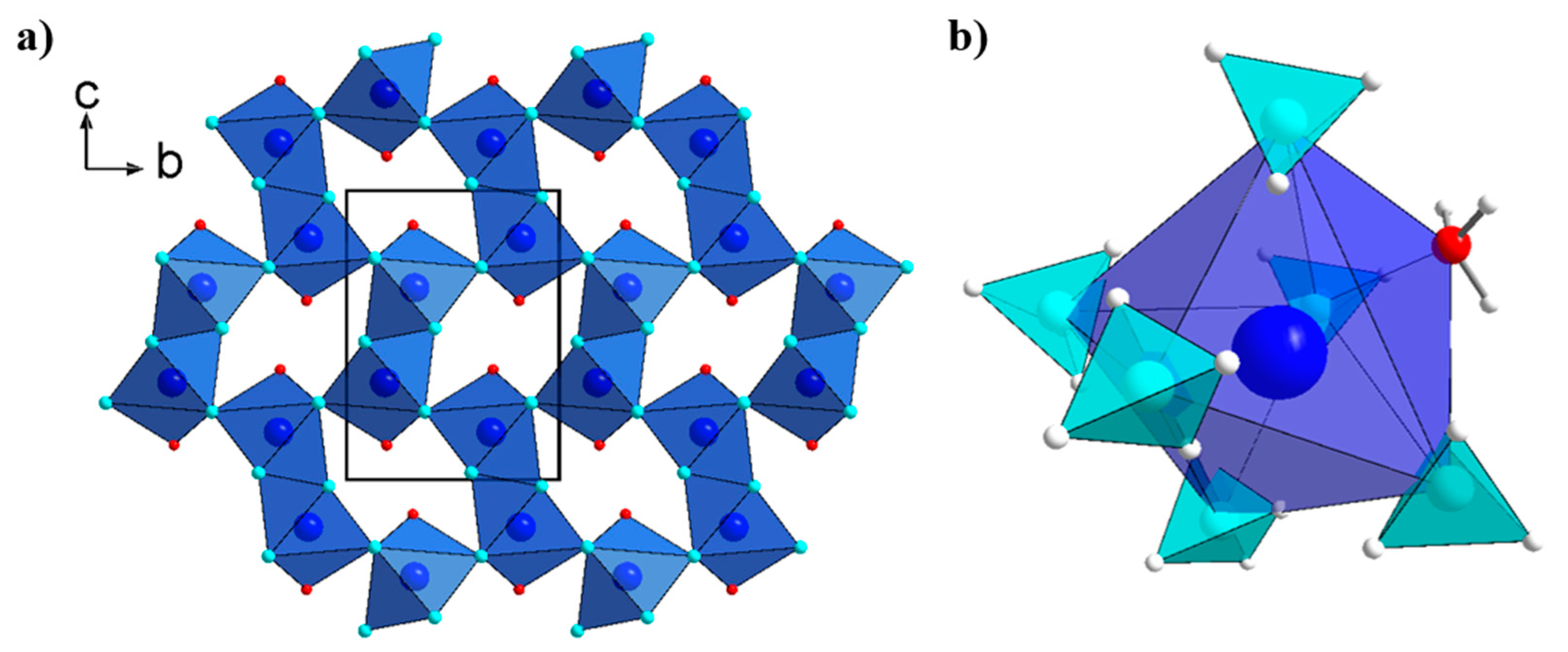
2.2. Crystal Structures
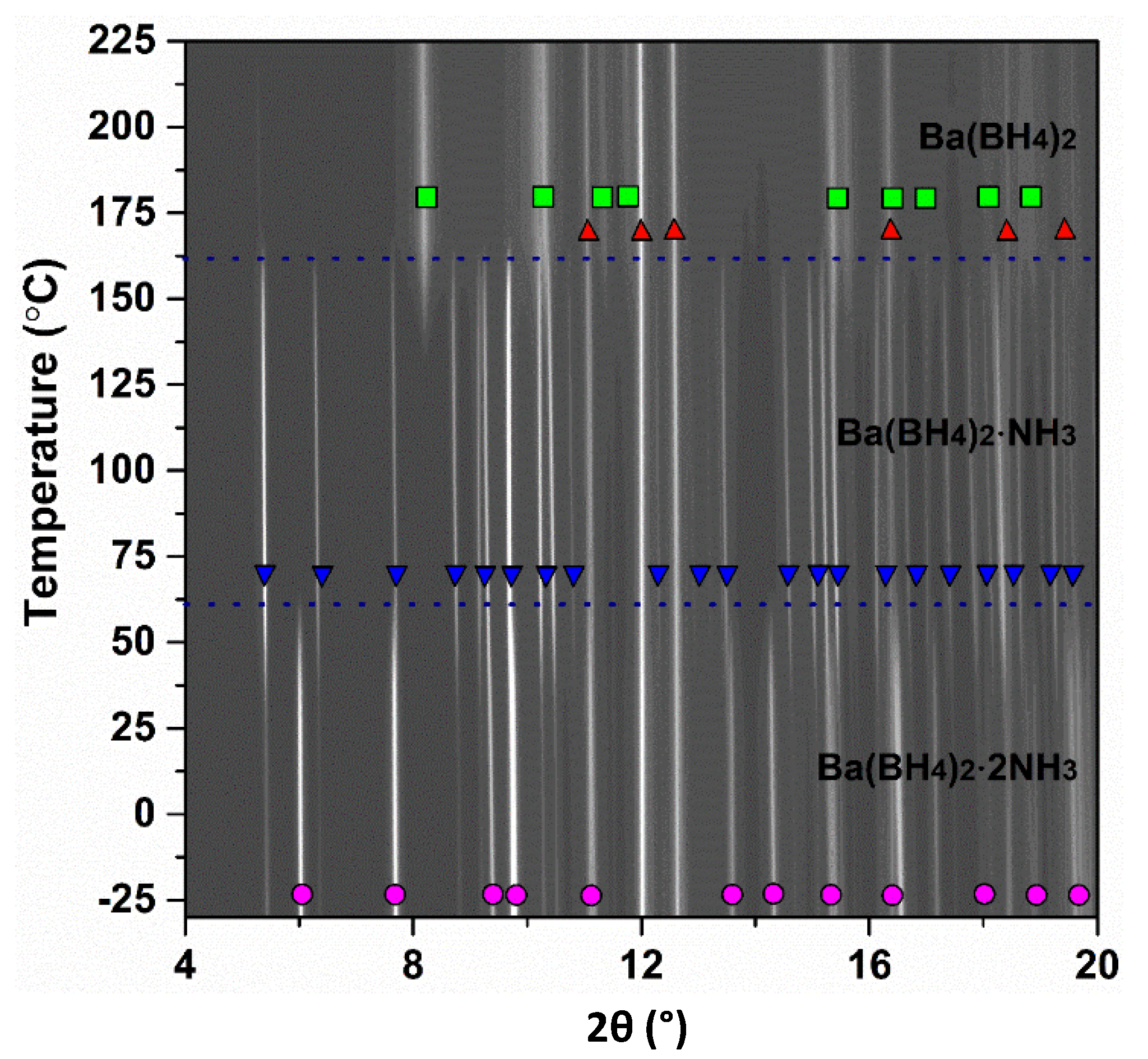
2.3. In Situ Synchrotron Powder X-ray Diffraction
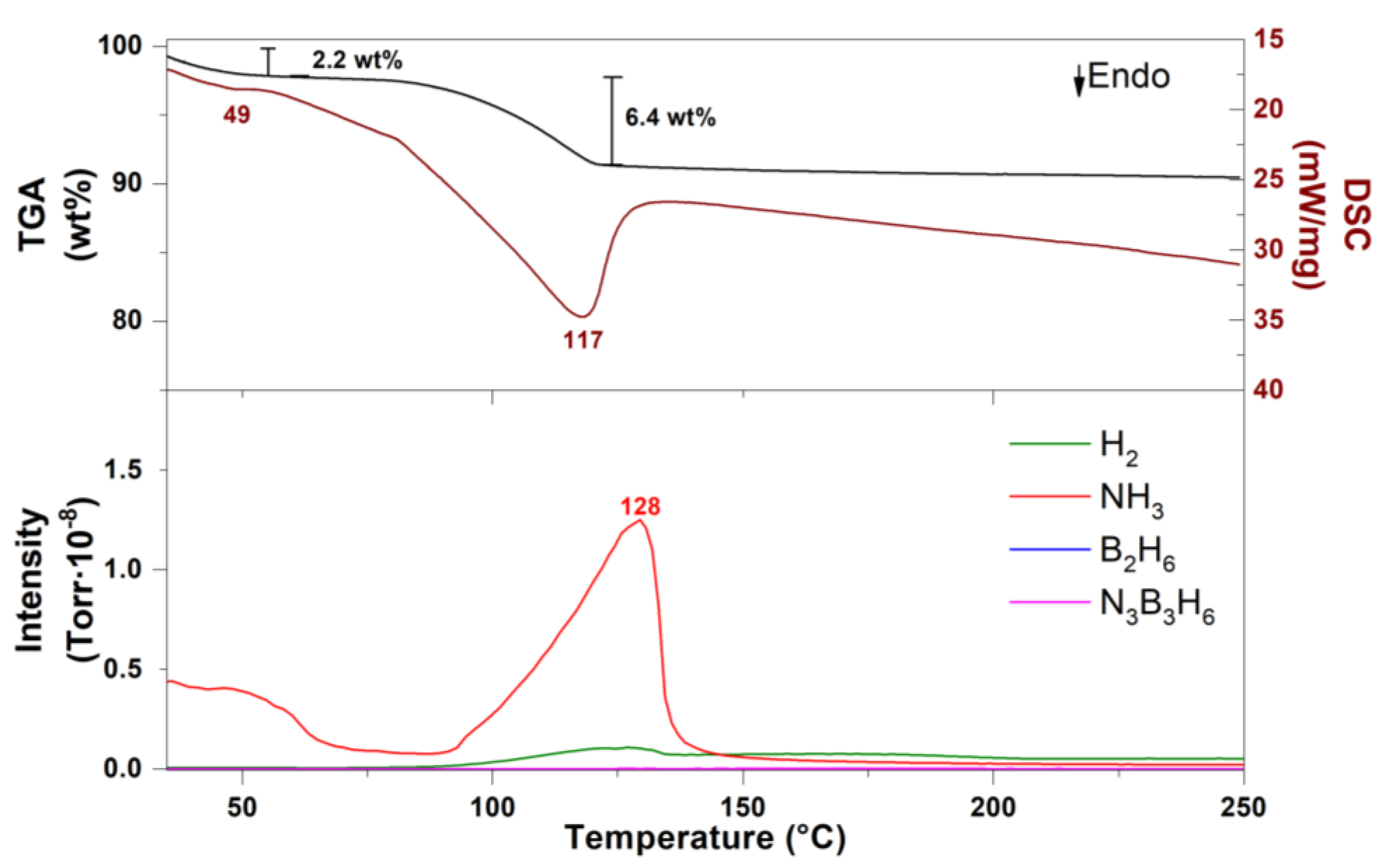
2.4. Thermal Analysis
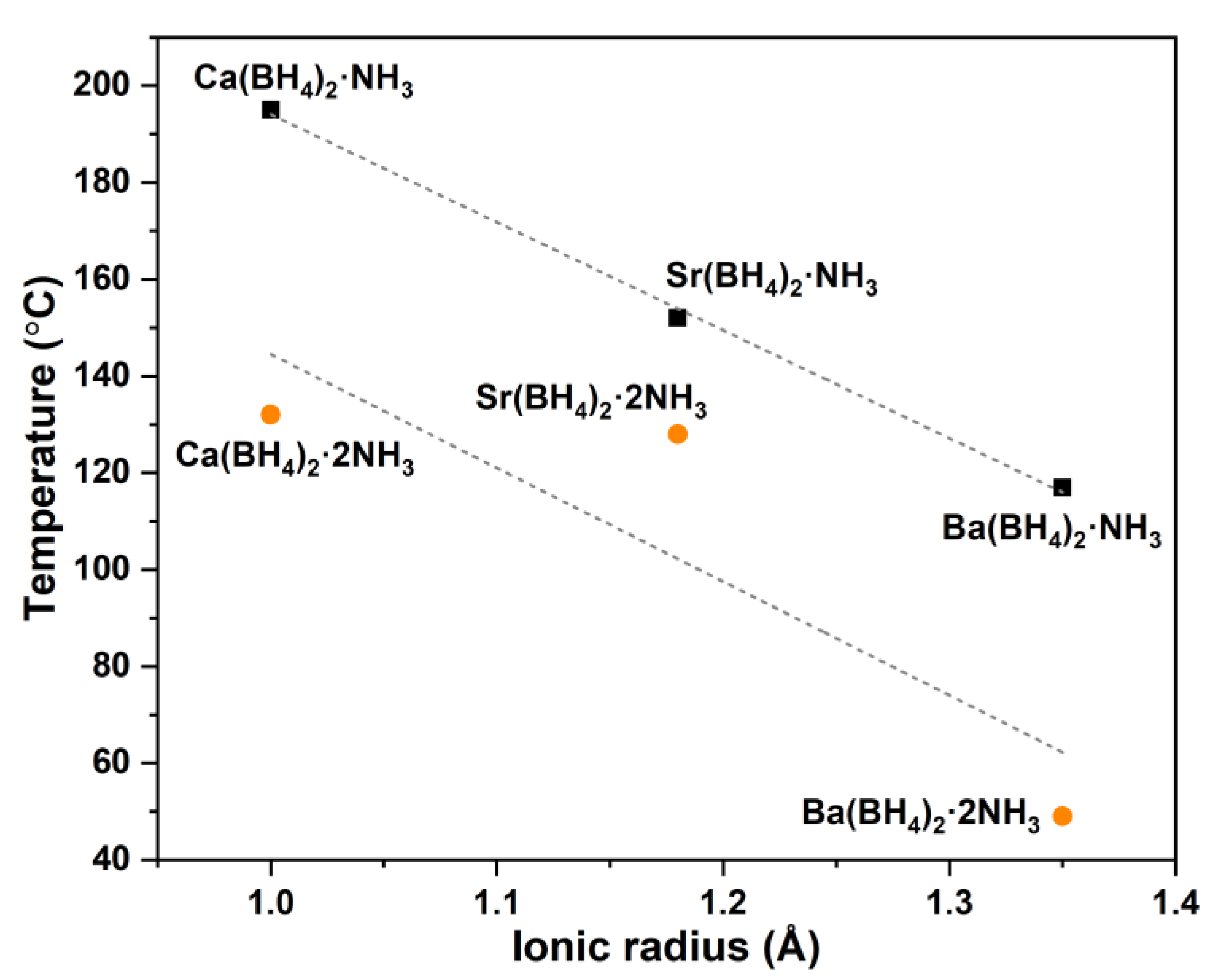
2.5. Mechanism for Thermal Decomposition
3. Materials and Methods
3.1. Sample Preparation
3.2. Synchrotron Radiation Powder X-ray Diffraction
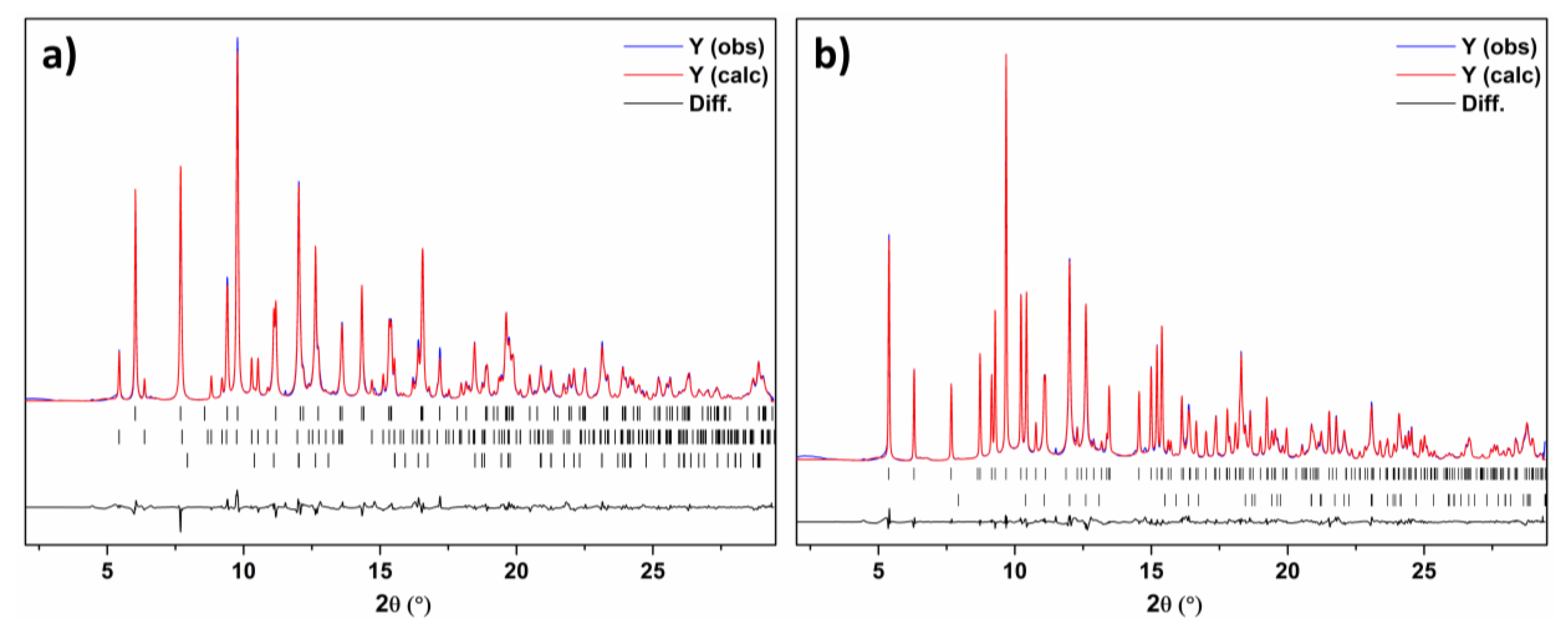
3.3. Structural Solution and Refinement
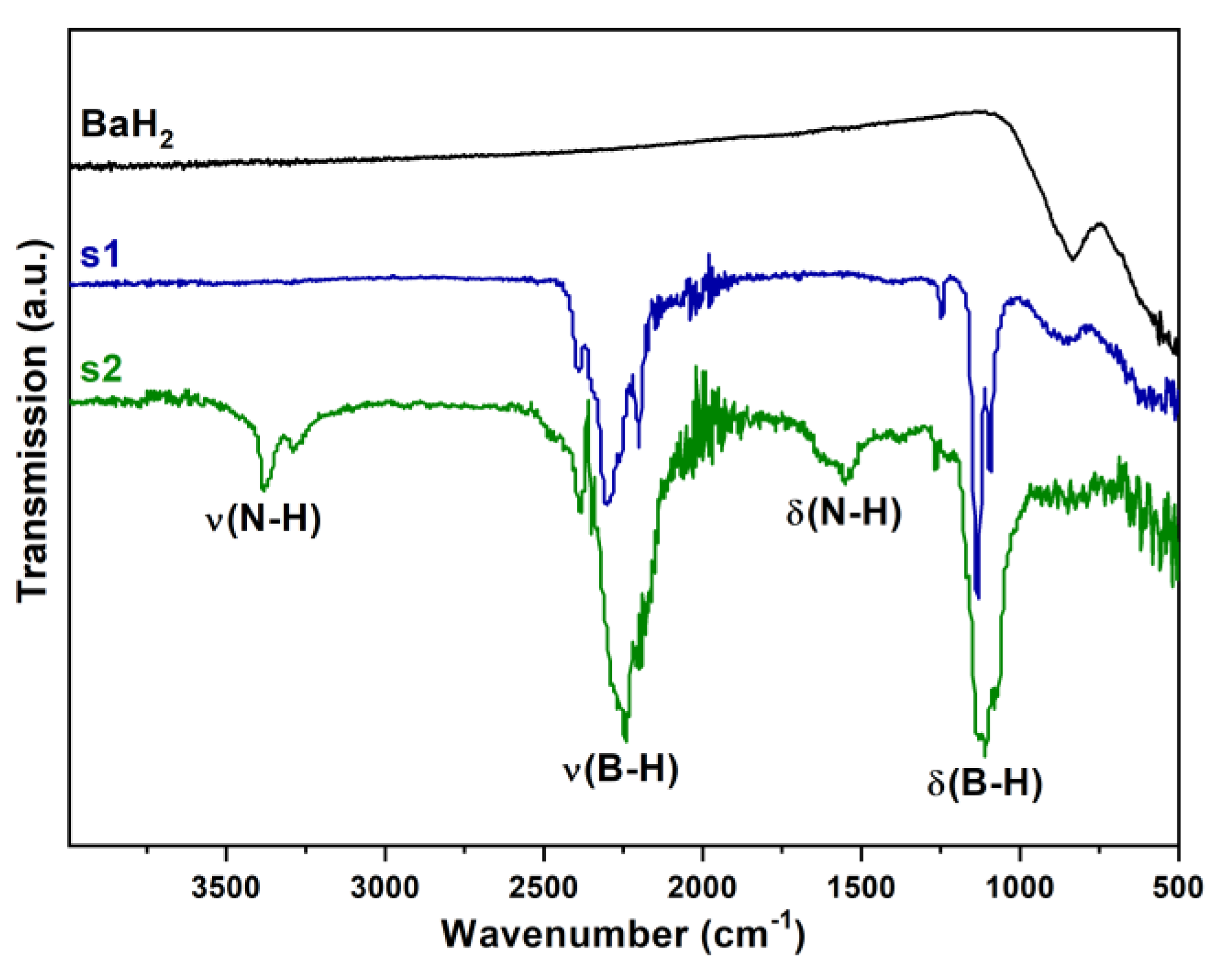
3.4. Fourier Transform Infrared Spectroscopy
3.5. Thermal Analysis and Mass Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Černý, R.; Ravnsbæk, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacher, F.; Jensen, T.R. Metal borohydrides and derivatives—Synthesis, structure and properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef] [PubMed]
- Møller, K.T.; Sheppard, D.; Ravnsbæk, D.B.; Buckley, C.E.; Akiba, E.; Li, H.W.; Jensen, T.R. Complex metal hydrides for hydrogen, thermal and electrochemical energy storage. Energies 2017, 10, 1645. [Google Scholar] [CrossRef] [Green Version]
- Hirscher, M.; Yartys, V.A.; Baricco, M.; Bellosta von Colbe, J.; Blanchard, D.; Bowman, R.C.; Broom, D.P.; Buckley, C.E.; Chang, F.; Chen, P.; et al. Materials for hydrogen-based energy storage—Past, recent progress and future outlook. J. Alloys Compd. 2020, 827, 153548. [Google Scholar] [CrossRef]
- Hadjixenophontos, E.; Dematteis, E.M.; Berti, N.; Wołczyk, A.R.; Huen, P.; Brighi, M.; Le, T.T.; Santoru, A.; Payandeh, S.; Peru, F.; et al. A Review of the MSCA ITN ECOSTORE—Novel Complex Metal Hydrides for Efficient and Compact Storage of Renewable Energy as Hydrogen and Electricity. Inorganics 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, H. Boron hydrogen compounds for hydrogen storage and as solid ionic conductors. Chimia 2019, 73, 868–873. [Google Scholar] [CrossRef]
- Schouwink, P.; Didelot, E.; Lee, Y.S.; Mazet, T.; Černý, R. Structural and magnetocaloric properties of novel gadolinium borohydrides. J. Alloys Compd. 2016, 664, 378–384. [Google Scholar] [CrossRef]
- Grinderslev, J.B.; Møller, K.T.; Bremholm, M.; Jensen, T.R. Trends in Synthesis, Crystal Structure, and Thermal and Magnetic Properties of Rare-Earth Metal Borohydrides. Inorg. Chem. 2019, 58, 5503–5517. [Google Scholar] [CrossRef]
- Wegner, W.; van Leusen, J.; Majewski, J.; Grochala, W.; Kögerler, P. Borohydride as Magnetic Superexchange Pathway in Late Lanthanide Borohydrides. Eur. J. Inorg. Chem. 2019, 2019, 1776–1783. [Google Scholar] [CrossRef]
- Marks, S.; Heck, J.G.; Habicht, M.H.; Oña-Burgos, P.; Feldmann, C.; Roesky, P.W. [Ln(BH4)2(THF)2] (Ln = Eu, Yb)—A highly luminescent material. Synthesis, properties, reactivity, and NMR studies. J. Am. Chem. Soc. 2012, 134, 16983–16986. [Google Scholar] [CrossRef]
- Schouwink, P.; Ley, M.B.; Tissot, A.; Hagemann, H.; Jensen, T.R.; Smrčok, Ľ.; Černý, R. Structure and properties of complex hydride perovskite materials. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Ley, M.B.; Ravnsbæk, D.B.; Filinchuk, Y.; Lee, Y.S.; Janot, R.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. LiCe(BH4)3Cl, a new lithium-ion conductor and hydrogen storage material with isolated tetranuclear anionic clusters. Chem. Mater. 2012, 24, 1654–1663. [Google Scholar] [CrossRef]
- de Jongh, P.E.; Blanchard, D.; Matsuo, M.; Udovic, T.J.; Orimo, S. Complex hydrides as room-temperature solid electrolytes for rechargeable batteries. Appl. Phys. A Mater. Sci. Process. 2016, 122, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Nakamori, Y.; Orimo, S.I.; Maekawa, H.; Takamura, H. Lithium superionic conduction in lithium borohydride accompanied by structural transition. Appl. Phys. Lett. 2007, 91, 224103. [Google Scholar] [CrossRef]
- Mohtadi, R.; Matsui, M.; Arthur, T.S.; Hwang, S.J. Magnesium borohydride: From hydrogen storage to magnesium battery. Angew. Chem. Int. Ed. 2012, 51, 9780–9783. [Google Scholar] [CrossRef] [PubMed]
- Heere, M.; Hansen, A.L.; Payandeh, S.H.; Aslan, N.; Gizer, G.; Sørby, M.H.; Hauback, B.C.; Pistidda, C.; Dornheim, M.; Lohstroh, W. Dynamics of porous and amorphous magnesium borohydride to understand solid state Mg-ion-conductors. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Makepeace, J.W.; He, T.; Weidenthaler, C.; Jensen, T.R.; Chang, F.; Vegge, T.; Ngene, P.; Kojima, Y.; de Jongh, P.E.; Chen, P.; et al. Reversible ammonia-based and liquid organic hydrogen carriers for high-density hydrogen storage: Recent progress. Int. J. Hydrogen Energy 2019, 44, 7746–7767. [Google Scholar] [CrossRef]
- Jepsen, L.H.; Ley, M.B.; Lee, Y.S.; Cho, Y.W.; Dornheim, M.; Jensen, J.O.; Filinchuk, Y.; Jørgensen, J.E.; Besenbacher, F.; Jensen, T.R. Boron-nitrogen based hydrides and reactive composites for hydrogen storage. Mater. Today 2014, 17, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, L.H.; Ley, M.B.; Černý, R.; Lee, Y.S.; Cho, Y.W.; Ravnsbæk, D.; Besenbacher, F.; Skibsted, J.; Jensen, T.R. Trends in Syntheses, Structures, and Properties for Three Series of Ammine Rare-Earth Metal Borohydrides, M(BH4)3·nNH3 (M = Y, Gd, and Dy). Inorg. Chem. 2015, 54, 7402–7414. [Google Scholar] [CrossRef]
- Soloveichik, G.; Her, J.H.; Stephens, P.W.; Gao, Y.; Rijssenbeek, J.; Andrus, M.; Zhao, J.C. Ammine magnesium borohydride complex as a new material for hydrogen storage: Structure and properties of Mg(BH4)2·2NH3. Inorg. Chem. 2008, 47, 4290–4298. [Google Scholar] [CrossRef]
- Filippov, S.; Grinderslev, J.B.; Andersson, M.S.; Armstrong, J.; Karlsson, M.; Jensen, T.R.; Klarbring, J.; Simak, S.I.; Häussermann, U. Analysis of Dihydrogen Bonding in Ammonium Borohydride. J. Phys. Chem. C 2019, 123, 28631–28639. [Google Scholar] [CrossRef]
- Grinderslev, J.B.; Jepsen, L.H.; Lee, Y.-S.; Møller, K.T.; Cho, Y.W.; Cerny, R.; Jensen, T.R. Structural Diversity and Trends in Properties of an Array of Hydrogen-Rich Ammonium Metal Borohydrides. Inorg. Chem. 2020, 59, 12733–12747. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Grinderslev, J.B.; Lee, Y.-S.; Jørgensen, M.; Cho, Y.W.; Černý, R.; Jensen, T.R. Ammonia-assisted fast Li-ion conductivity in a new hemiammine lithium borohydride, LiBH4·½NH3. Chem. Commun. 2020, 56, 3971–3974. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Dononelli, W.; Jørgensen, M.; Grinderslev, J.B.; Lee, Y.-S.; Cho, Y.W.; Černý, R.; Hammer, B.; Jensen, T.R. The mechanism of Mg2+ conduction in ammine magnesium borohydride promoted by a neutral molecule. Phys. Chem. Chem. Phys. 2020, 22, 9204–9209. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Grinderslev, J.B.; Jørgensen, M.; Skov, L.N.; Skibsted, J.; Jensen, T.R. Ammine Magnesium Borohydride Nanocomposites for All-Solid-State Magnesium Batteries. ACS Appl. Energy Mater. 2020, 3, 9264–9270. [Google Scholar] [CrossRef]
- Jepsen, L.H.; Lee, Y.S.; Černý, R.; Sarusie, R.S.; Cho, Y.W.; Besenbacher, F.; Jensen, T.R. Ammine Calcium and Strontium Borohydrides: Syntheses, Structures, and Properties. ChemSusChem 2015, 8, 3472–3482. [Google Scholar] [CrossRef] [PubMed]
- Grinderslev, J.B.; Ley, M.B.; Lee, Y.-S.; Jepsen, L.H.; Jørgensen, M.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. Ammine Lanthanum and Cerium Borohydrides, M(BH4)3·nNH3; Trends in Synthesis, Structures, and Thermal Properties. Inorg. Chem. 2020, 59, 7768–7778. [Google Scholar] [CrossRef]
- Richter, B.; Ravnsbæk, D.B.; Tumanov, N.; Filinchuk, Y.; Jensen, T.R. Manganese borohydride; Synthesis and characterization. Dalt. Trans. 2015, 44, 3988–3996. [Google Scholar] [CrossRef]
- Yan, Y.; Li, H.W.; Sato, T.; Umeda, N.; Miwa, K.; Towata, S.; Orimo, S. Dehydriding and rehydriding properties of yttrium borohydride Y(BH4)3 prepared by liquid-phase synthesis. Int. J. Hydrogen Energy 2009, 34, 5732–5736. [Google Scholar] [CrossRef]
- Rude, L.H.; Corno, M.; Ugliengo, P.; Baricco, M.; Lee, Y.S.; Cho, Y.W.; Besenbacher, F.; Overgaard, J.; Jensen, T.R. Synthesis and structural investigation of Zr(BH4)4. J. Phys. Chem. C 2012, 116, 20239–20245. [Google Scholar] [CrossRef]
- Ravnsbœk, D.B.; Sørensen, L.H.; Filinchuk, Y.; Besenbacher, F.; Jensen, T.R. Screening of metal borohydrides by mechanochemistry and diffraction. Angew. Chem. Int. Ed. 2012, 51, 3582–3586. [Google Scholar] [CrossRef]
- Li, S.; Yang, B.; Tang, S.; Lai, T.; Xiang, J.; Wang, L. Chemical synthesis and characterization of zinc borohydride. Procedia Eng. 2012, 27, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Roedern, E.; Jensen, T.R. Ammine-Stabilized Transition-Metal Borohydrides of Iron, Cobalt, and Chromium: Synthesis and Characterization. Inorg. Chem. 2015, 54, 10477–10482. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, L.H.; Ley, M.B.; Filinchuk, Y.; Besenbacher, F.; Jensen, T.R. Tailoring the Properties of Ammine Metal Borohydrides for Solid-State Hydrogen Storage. ChemSusChem 2015, 8, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tan, Y.; Su, J.; Gu, Q.; Černý, R.; Ouyang, L.; Sun, D.; Yu, X.; Zhu, M. Synthesis, structure and dehydrogenation of zirconium borohydride octaammoniate. Chem. Commun. 2015, 51, 2794–2797. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, X.; Gu, Q.; Tang, Z.; Yu, X. Synthesis of ammine dual-metal (V, Mg) borohydrides with enhanced dehydrogenation properties. Int. J. Hydrogen Energy 2013, 38, 5322–5329. [Google Scholar] [CrossRef]
- Yuan, F.; Gu, Q.; Chen, X.; Tan, Y.; Guo, Y.; Yu, X. Complex ammine titanium(III) borohydrides as advanced solid hydrogen-storage materials with favorable dehydrogenation properties. Chem. Mater. 2012, 24, 3370–3379. [Google Scholar] [CrossRef]
- Welchman, E.; Thonhauser, T. Decomposition mechanisms in metal borohydrides and their ammoniates. J. Mater. Chem. A 2017, 5, 4084–4092. [Google Scholar] [CrossRef] [Green Version]
- Marynick, D.S.; Lipscomb, W.N. Crystal Structure of Beryllium Borohydride. Inorg. Chem. 1972, 11, 820–823. [Google Scholar] [CrossRef]
- Sharma, M.; Didelot, E.; Spyratou, A.; Max Lawson Daku, L.; Černý, R.; Hagemann, H. Halide Free M(BH4)2 (M = Sr, Ba, and Eu) Synthesis, Structure, and Decomposition. Inorg. Chem. 2016, 55, 7090–7097. [Google Scholar] [CrossRef]
- D’Anna, V.; Spyratou, A.; Sharma, M.; Hagemann, H. FT-IR spectra of inorganic borohydrides. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 128, 902–906. [Google Scholar] [CrossRef]
- Olovsson, I.; Templeton, D.H. X-ray study of solid ammonia. Acta Cryst. 1959, 12, 832–836. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Tang, Z.; Tan, Y.; Gu, Q.; Yu, X. A novel aided-cation strategy to advance the dehydrogenation of calcium borohydride monoammoniate. J. Mater. Chem. 2012, 22, 5312–5318. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Li, Y.; Gao, M.; Pan, H. Synthesis and thermal decomposition behaviors of magnesium borohydride ammoniates with controllable composition as hydrogen storage materials. Chem. Asian J. 2013, 8, 476–481. [Google Scholar] [CrossRef]
- Richter, B.; Grinderslev, J.B.; Møller, K.T.; Paskevicius, M.; Jensen, T.R. From Metal Hydrides to Metal Borohydrides. Inorg. Chem. 2018, 57, 10768–10780. [Google Scholar] [CrossRef] [PubMed]
- Dyadkin, V.; Pattison, P.; Dmitriev, V.; Chernyshov, D. A new multipurpose diffractometer PILATUS@SNBL. J. Synchrotron Radiat. 2016, 23, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Favre-Nicolin, V.; Černý, R. FOX, “free objects for crystallography”: A modular approach to ab initio structure determination from powder diffraction. J. Appl. Cryst. 2002, 35, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Carvajal, R.J. FULLPROF: A Program for Rietveld Refinement and Pattern Matching Analysis; Abstracts of the Satellite Meeting on Powder Diffraction of the XV Congress of the IUCr; International Union of Crystallography: Toulouse, France, 1990; p. 127. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. Sect. D Biol. Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Reactants | Products after Synthesis |
|---|---|---|
| s1 | BaH2 + DMS-BH3 | Ba(BH4)2 + BaH2 |
| s2 | Ba(BH4)2 + NH3 (+ BaH2) | Ba(BH4)2·2NH3 + Ba(BH4)2·NH3 + BaH2 |
| Ba(BH4)2·2NH3 | Ba(BH4)2·NH3 | |
|---|---|---|
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Pnc2 | P212121 |
| T (°C) * | −25 | 95 |
| a (Å) | 6.7784(2) | 5.08626(8) |
| b (Å) | 6.7175(2) | 9.3761(2) |
| c (Å) | 8.6991(3) | 12.9632(2) |
| V (Å3) | 396.10(2) | 618.20(2) |
| Z | 2 | 4 |
| V/Z (Å3) | 198.1 | 154.6 |
| M (g/mol) | 201.08 | 184.05 |
| ρ (g/mL) | 1.686 | 1.977 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinderslev, J.B.; Amdisen, M.B.; Jensen, T.R. Synthesis, Crystal Structures and Thermal Properties of Ammine Barium Borohydrides. Inorganics 2020, 8, 57. https://doi.org/10.3390/inorganics8100057
Grinderslev JB, Amdisen MB, Jensen TR. Synthesis, Crystal Structures and Thermal Properties of Ammine Barium Borohydrides. Inorganics. 2020; 8(10):57. https://doi.org/10.3390/inorganics8100057
Chicago/Turabian StyleGrinderslev, Jakob B., Mads B. Amdisen, and Torben R. Jensen. 2020. "Synthesis, Crystal Structures and Thermal Properties of Ammine Barium Borohydrides" Inorganics 8, no. 10: 57. https://doi.org/10.3390/inorganics8100057
APA StyleGrinderslev, J. B., Amdisen, M. B., & Jensen, T. R. (2020). Synthesis, Crystal Structures and Thermal Properties of Ammine Barium Borohydrides. Inorganics, 8(10), 57. https://doi.org/10.3390/inorganics8100057