
Hydrothermal Synthesis and Structural Investigation of a Crystalline Uranyl Borosilicate

 , , , and
, , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Synthesis
2.3. Single Crystal X-ray Diffraction
2.4. Topological Analysis
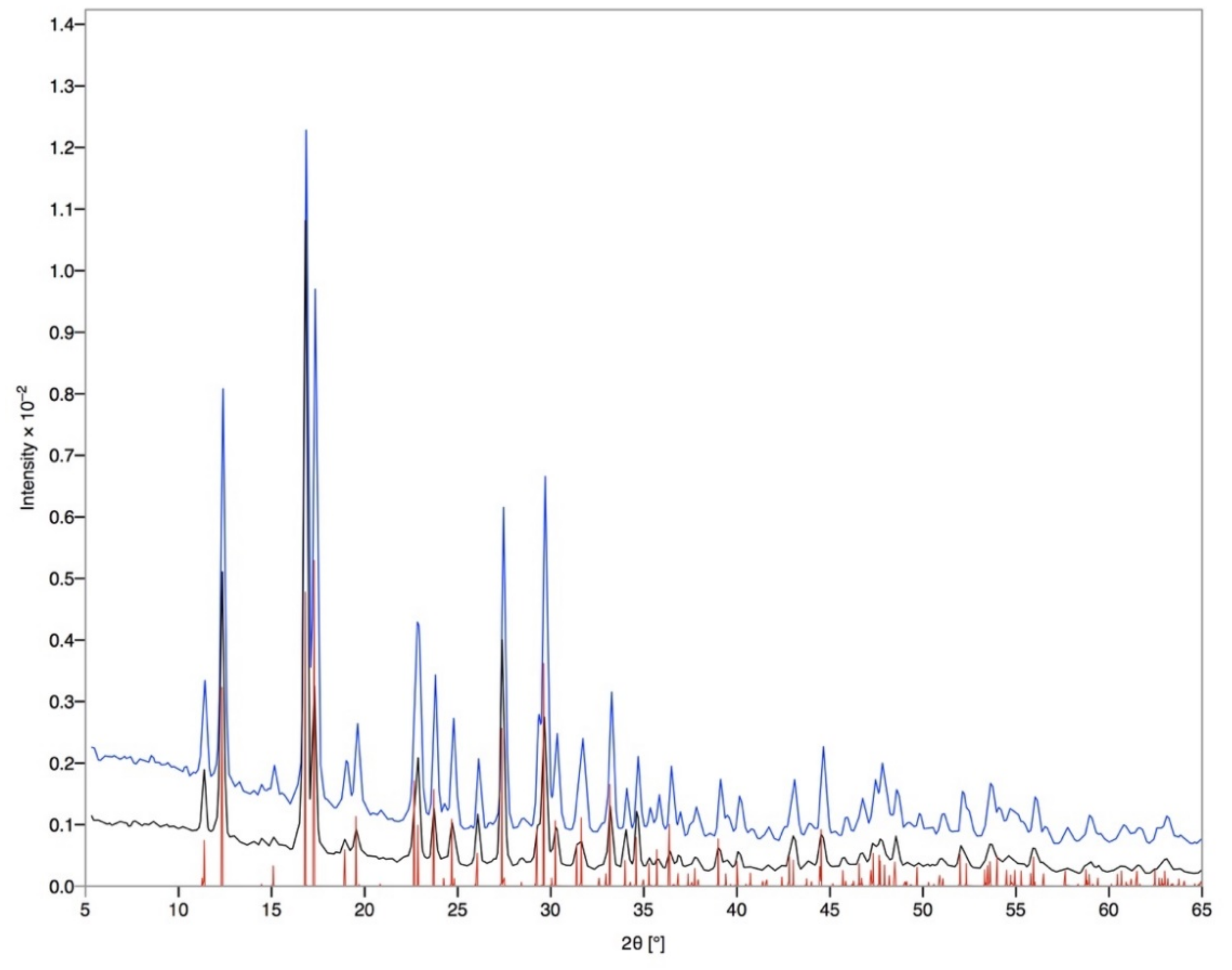
2.5. Powder X-ray Diffraction
2.6. Spectroscopy
2.7. Thermal Properties
2.8. First Principles Calculations
3. Results and Discussion
3.1. Synthesis
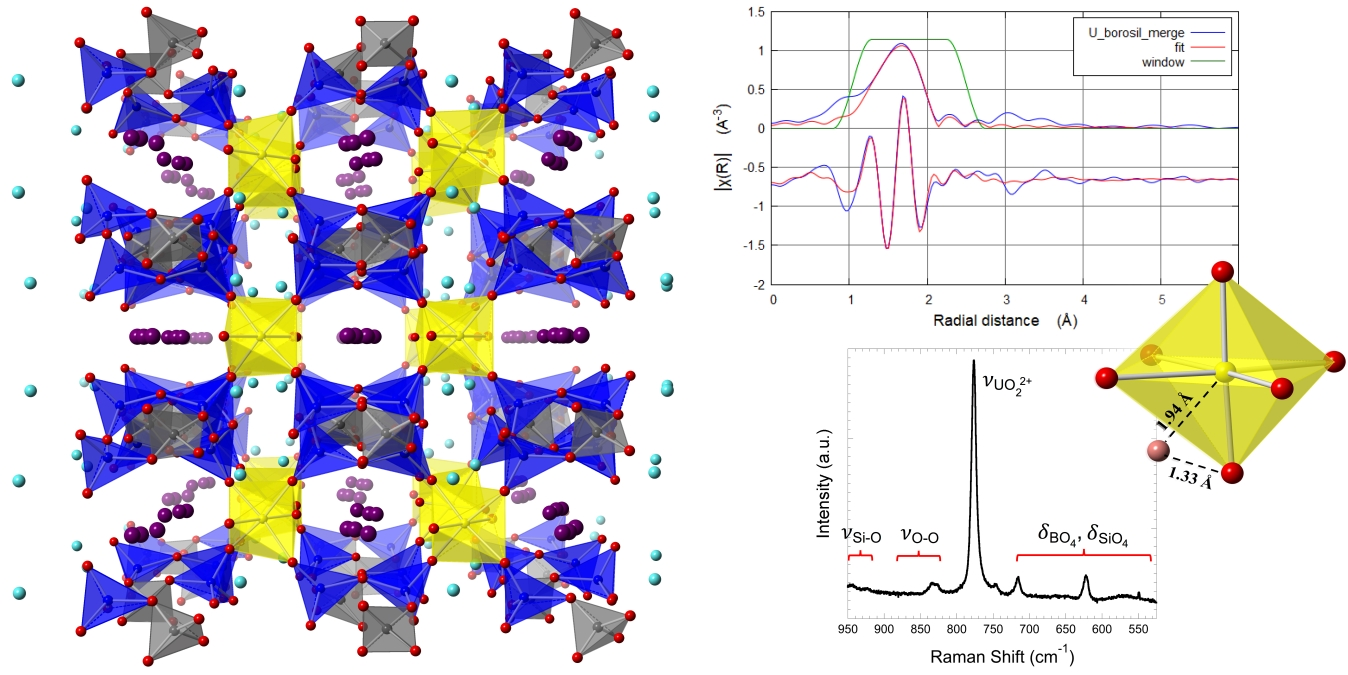
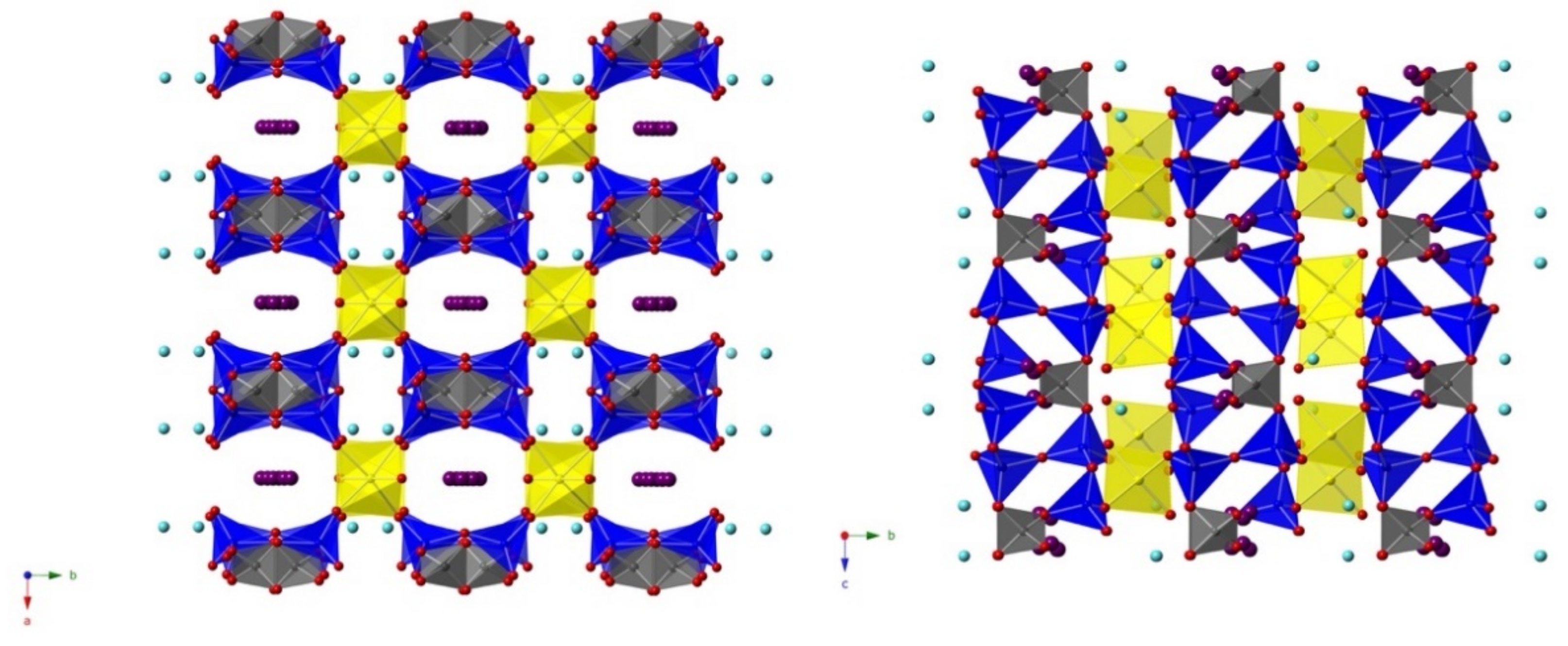
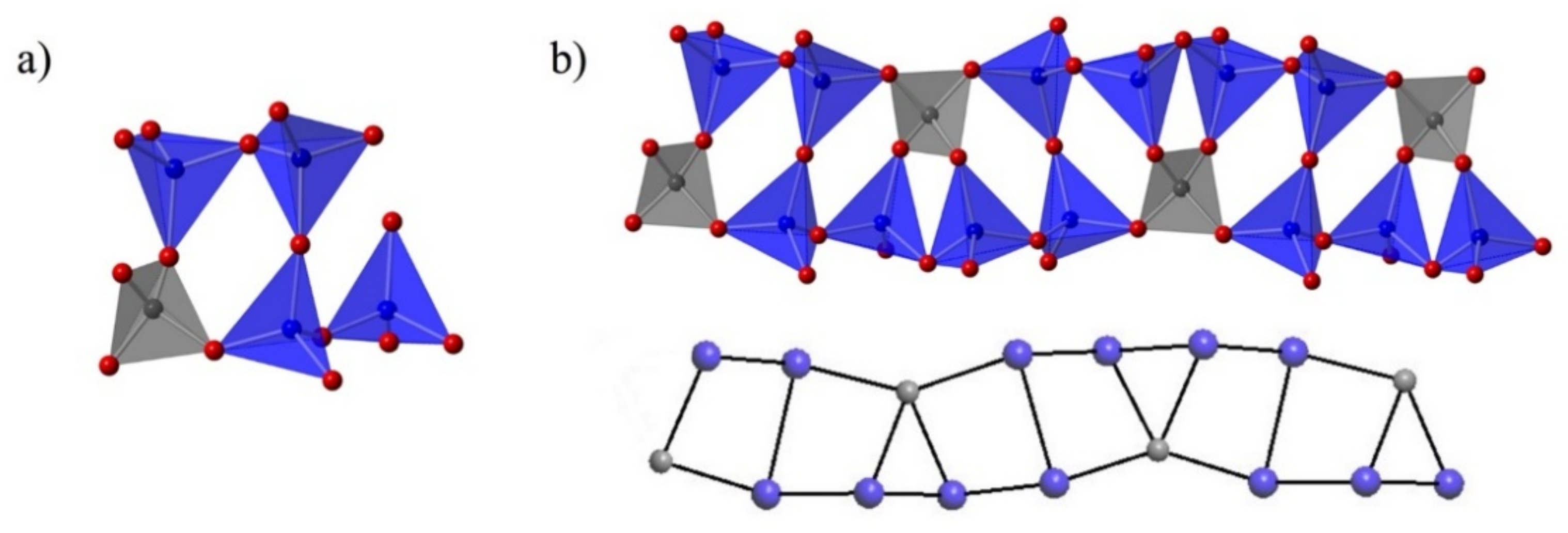
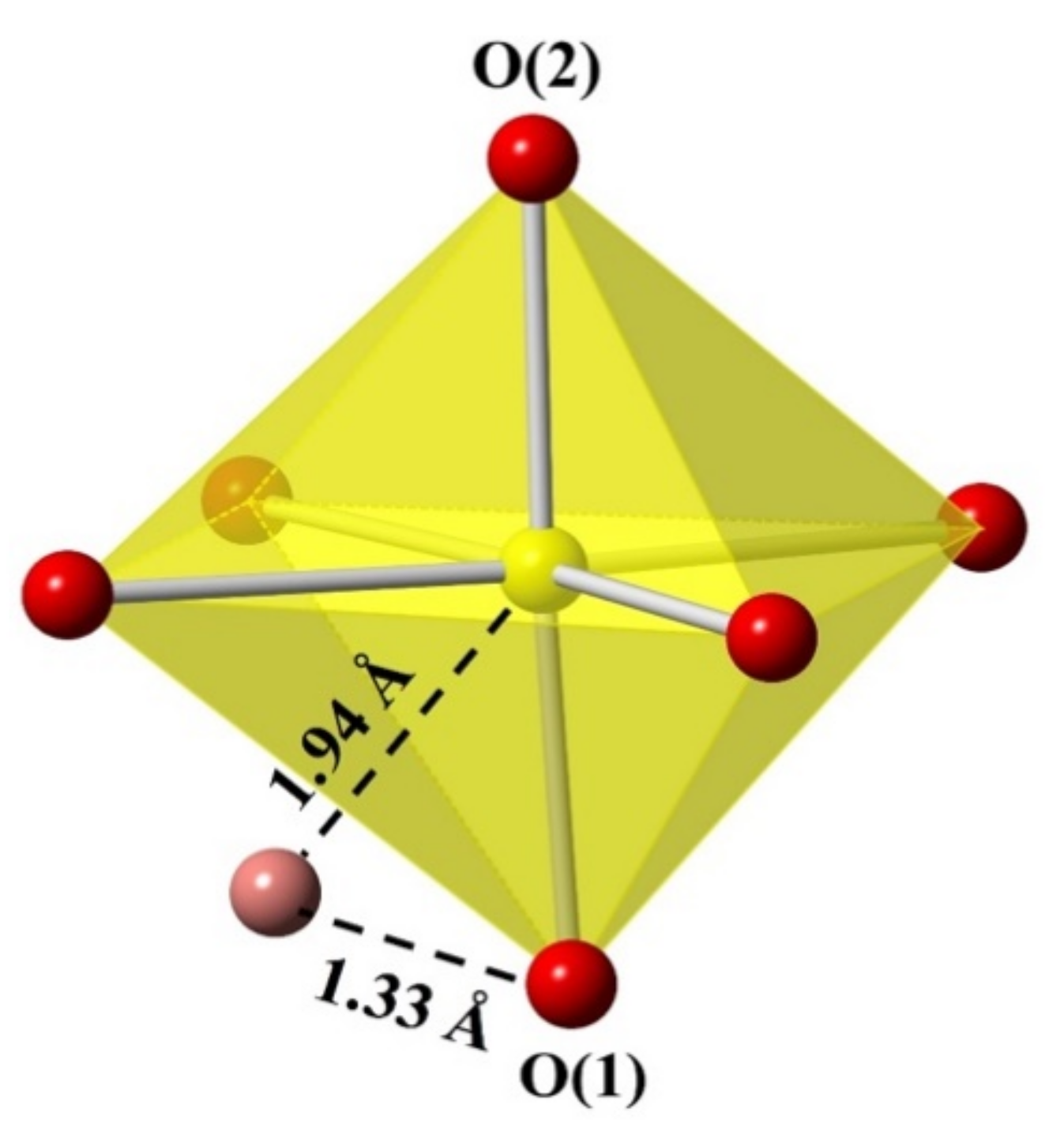
3.2. Structure Description
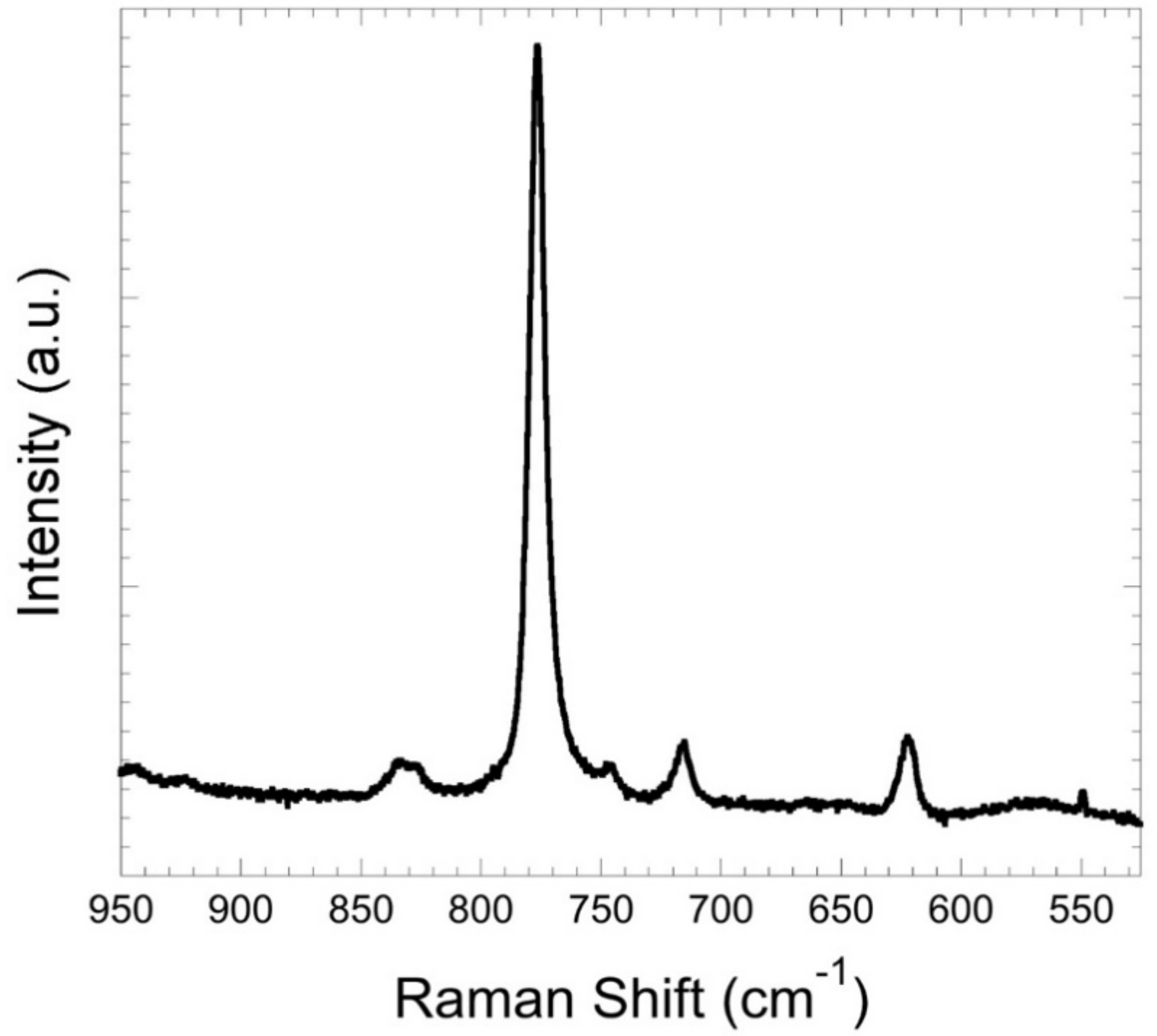
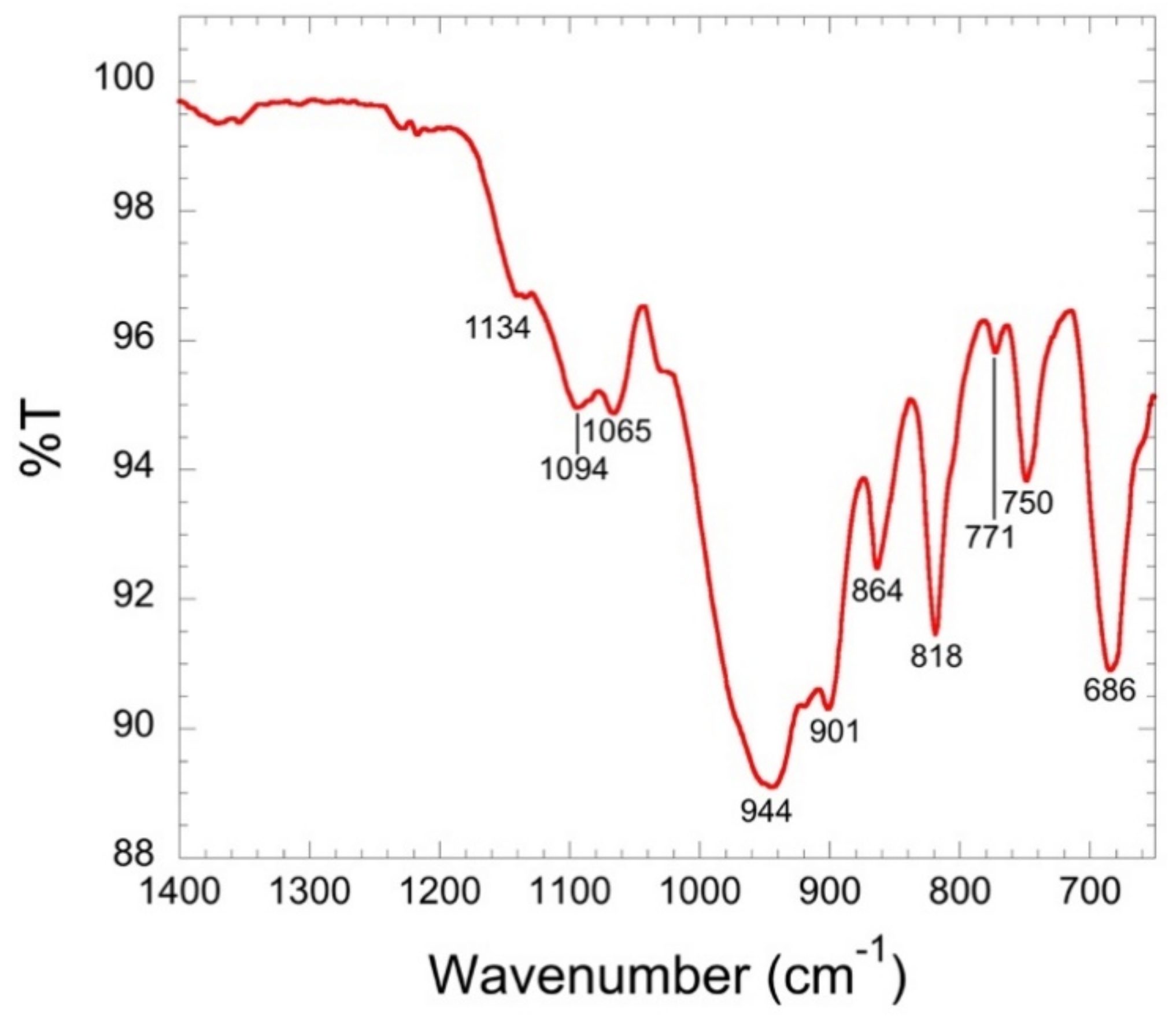
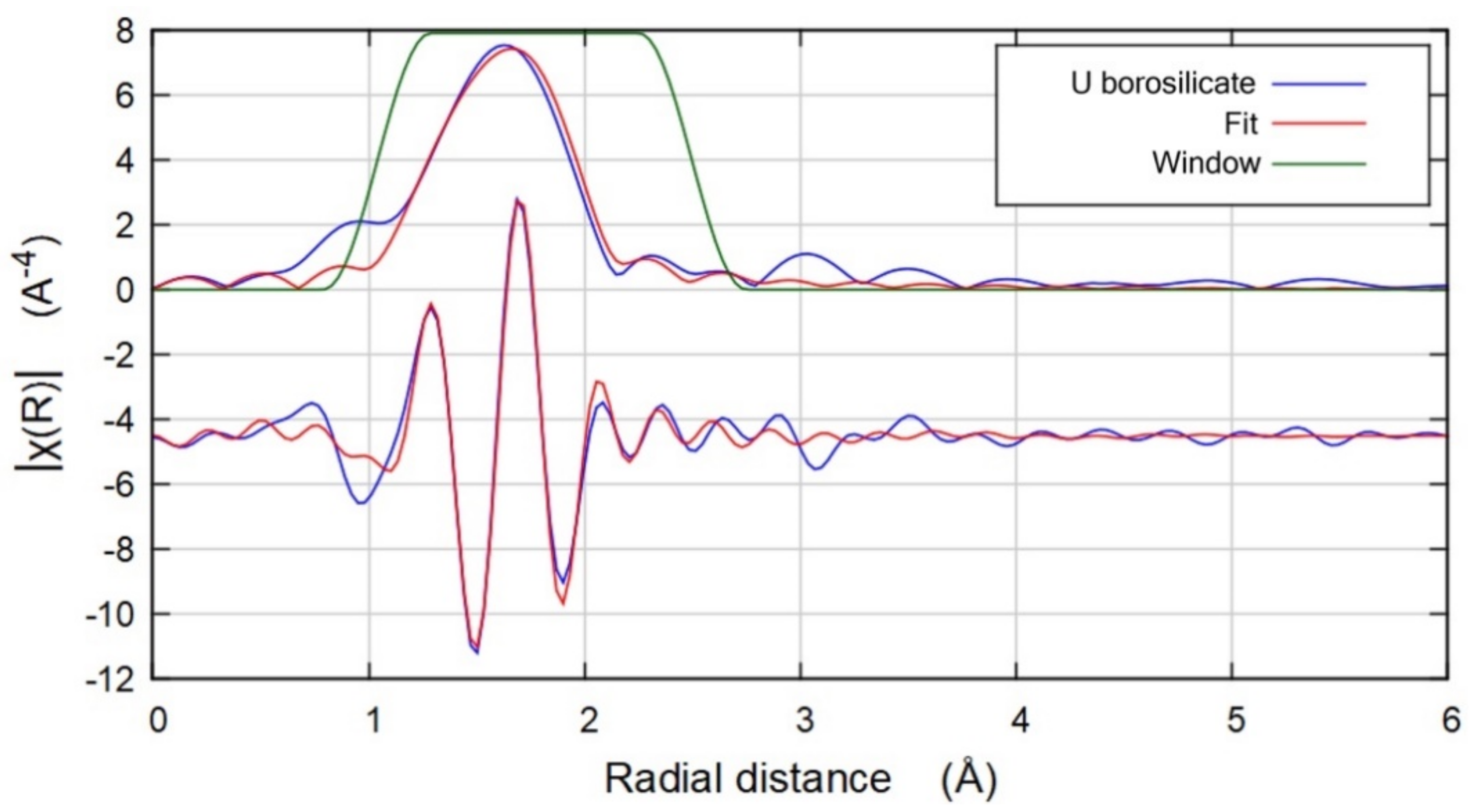
3.3. Spectroscopy
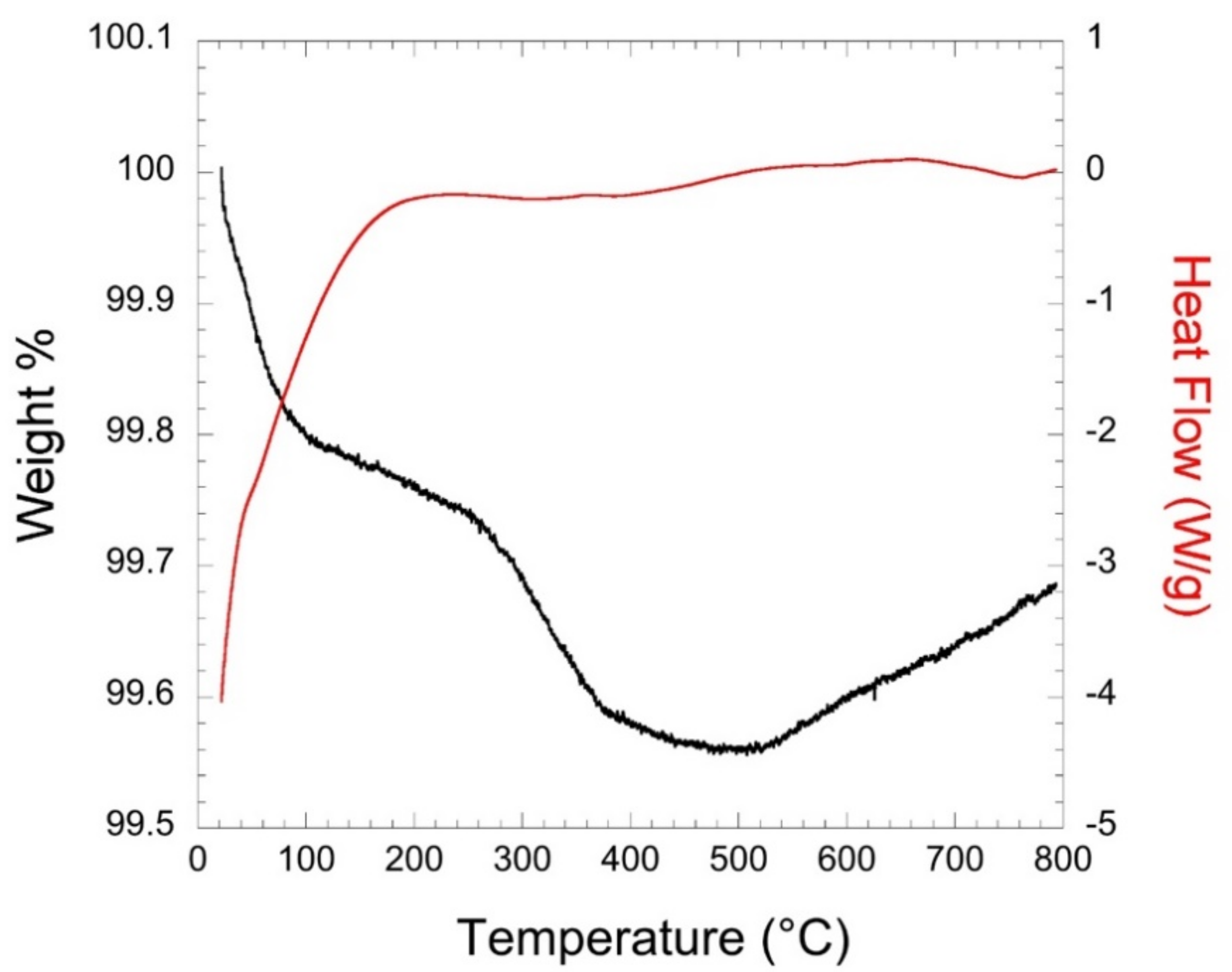
3.4. Thermal Properties
3.5. Energy Optimization Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Research Council. Waste Forms Technology and Performance; Final Report by National Research Council of the National Academies; The National Academies Press: Washington, DC, USA, 2011.
- Burns, P.C.; Olson, R.A.; Finch, R.J.; Hanchar, J.M.; Thibault, Y. KNa3(UO2)2(Si4O10)2(H2O)4, a new compound formed during vapor hydration of an actinide-bearing borosilicate waste glass. J. Nucl. Mater. 2000, 278, 290–300. [Google Scholar] [CrossRef]
- Wang, S.; Alekseev, E.V.; Ling, J.; Skanthakumar, S.; Soderholm, L.; Depmeier, W.; Albrecht-Schmitt, T.E. Neptunium diverges sharply from uranium and plutonium in crystalline borate matrixes: Insights into the complex behavior of the early actinides relevant to nuclear waste storage. Angew. Chem. Int. Ed. Engl. 2010, 49, 1263–1266. [Google Scholar] [CrossRef]
- Weber, W.J.; Navrotsky, A.; Stefanovsky, S.V.; Vance, E.R.; Vernaz, E.Y. Materials science of high-level nuclear waste immobilization. MRS Bull. 2009, 34, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Zur Loye, H.-C.; Besmann, T.; Amoroso, J.; Brinkman, K.; Grandjean, A.; Henager, C.H.; Hu, S.; Misture, S.T.; Phillpot, S.R.; Shustova, N.B.; et al. Hierarchical Materials as Tailored Nuclear Waste Forms: A Perspective. Chem. Mater. 2018, 30, 4475–4488. [Google Scholar] [CrossRef]
- Xu, Y.; Wen, Y.; Grote, R.; Amoroso, J.; Shuller Nickles, L.; Brinkman, K.S. A-site compositional effects in Ga-doped hollandite materials of the form BaxCsyGa2x+yTi8−2x−yO16: Implications for Cs immobilization in crystalline ceramic waste forms. Sci. Rep. 2016, 6, 27412. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Alekseev, E.; Diwu, J.; Casey, W.; Phillips, B.; Depmeier, W.; Albrecht-Schmitt, T. NDTB-1: A Supertetrahedral Cationic Framework That Removes TcO4− from Solution. Angew. Chem. Int. Ed. 2010, 49, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Halasyamani, P.S.; Walker, S.M.; O’Hare, D. The First Open Framework Actinide Material (C4N2H12)U2O4F6 (MUF-1). J. Am. Chem. Soc. 1999, 121, 7415–7416. [Google Scholar] [CrossRef]
- Jackson, J.M.; Burns, P.C. A re-evaluation of the structure of weeksite, a uranyl silicate framework mineral. Can. Mineral. 2001, 39, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, J.; Liu, L.; Jacobson, A.J. The novel open-framework uranium silicates Na2(UO2)(Si4O10)·2.1H2O (USH-1) and RbNa(UO2)(Si2O6)·H2O (USH-3). J. Mater. Chem. 2002, 12, 406–410. [Google Scholar] [CrossRef]
- Chen, C.-S.; Kao, H.-M.; Lii, K.-H. K5(UO2)2[Si4O12(OH)]: A uranyl silicate containing chains of four silicate tetrahedra linked by SiO···HOSi hydrogen bonds. Inorg. Chem. 2005, 44, 935–940. [Google Scholar] [CrossRef]
- Liu, H.-K.; Chang, W.-J.; Lii, K.-H. High-Temperature, High-Pressure Hydrothermal Synthesis and Characterization of an Open-Framework Uranyl Silicate with Nine-Ring Channels: Cs2UO2Si10O22. Inorg. Chem. 2011, 50, 11773–11776. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-K.; Peng, C.-C.; Chang, W.-J.; Lii, K.-H. Tubular Chains, Single Layers, and Multiple Chains in Uranyl Silicates: A2[(UO2)Si4O10] (A = Na, K, Rb, Cs). Cryst. Growth Des. 2016, 16, 5268–5272. [Google Scholar] [CrossRef]
- Morrison, G.; Tran, T.T.; Halasyamani, P.S.; zur Loye, H.-C. K8(K5F)U6Si8O40: An Intergrowth Uranyl Silicate. Inorg. Chem. 2016, 55, 3215–3217. [Google Scholar] [CrossRef]
- Morrison, G.; Smith, M.D.; zur Loye, H.-C. Understanding the Formation of Salt-Inclusion Phases: An Enhanced Flux Growth Method for the Targeted Synthesis of Salt-Inclusion Cesium Halide Uranyl Silicates. J. Am. Chem. Soc. 2016, 138, 7121–7129. [Google Scholar] [CrossRef]
- Li, H.; Kegler, P.; Bosbach, D.; Alekseev, E.V. Hydrothermal Synthesis, Study, and Classification of Microporous Uranium Silicates and Germanates. Inorg. Chem. 2018, 57, 4745–4756. [Google Scholar] [CrossRef]
- Chen, C.-S.; Lee, S.-F.; Lii, K.-H. K(UO)Si2O6: A pentavalent-uranium silicate. J. Am. Chem. Soc. 2005, 127, 12208–12209. [Google Scholar] [CrossRef]
- Wang, S.; Alekseev, E.V.; Stritzinger, J.T.; Depmeier, W.; Albrecht-Schmitt, T.E. Crystal chemistry of the potassium and rubidium uranyl borate families derived from boric acid fluxes. Inorg. Chem. 2010, 49, 6690–6696. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Alekseev, E.V.; Stritzinger, J.T.; Depmeier, W.; Albrecht-Schmitt, T.E. How are centrosymmetric and noncentrosymmetric structures achieved in uranyl borates? Inorg. Chem. 2010, 49, 2948–2953. [Google Scholar] [CrossRef]
- Wang, S.; Alekseev, E.V.; Ling, J.; Liu, G.; Depmeier, W.; Albrecht-Schmitt, T.E. Polarity and Chirality in Uranyl Borates: Insights into Understanding the Vitrification of Nuclear Waste and the Development of Nonlinear Optical Materials. Chem. Mater. 2010, 22, 2155–2163. [Google Scholar] [CrossRef]
- Wang, S.; Alekseev, E.V.; Stritzinger, J.T.; Liu, G.; Depmeier, W.; Albrecht-Schmitt, T.E. Structure–Property Relationships in Lithium, Silver, and Cesium Uranyl Borates. Chem. Mater. 2010, 22, 5983–5991. [Google Scholar] [CrossRef]
- Wu, S.; Wang, S.; Polinski, M.; Beermann, O.; Kegler, P.; Malcherek, T.; Holzheid, A.; Depmeier, W.; Bosbach, D.; Albrecht-Schmitt, T.E.; et al. High structural complexity of potassium uranyl borates derived from high-temperature/high-pressure reactions. Inorg. Chem. 2013, 52, 5110–5118. [Google Scholar] [CrossRef]
- Hao, Y.; Klepov, V.V.; Murphy, G.L.; Modolo, G.; Bosbach, D.; Albrecht-Schmitt, T.E.; Kennedy, B.J.; Wang, S.; Alekseev, E.V. Influence of Synthetic Conditions on Chemistry and Structural Properties of Alkaline Earth Uranyl Borates. Cryst. Growth Des. 2016, 16, 5923–5931. [Google Scholar] [CrossRef]
- Hao, Y.; Klepov, V.V.; Kegler, P.; Modolo, G.; Bosbach, D.; Albrecht-Schmitt, T.E.; Wang, S.; Alekseev, E.V. Synthesis and Study of the First Zeolitic Uranium Borate. Cryst. Growth Des. 2018, 18, 498–505. [Google Scholar] [CrossRef]
- Wu, S.; Polinski, M.J.; Malcherek, T.; Bismayer, U.; Klinkenberg, M.; Modolo, G.; Bosbach, D.; Depmeier, W.; Albrecht-Schmitt, T.E.; Alekseev, E.V. Novel fundamental building blocks and site dependent isomorphism in the first actinide borophosphates. Inorg. Chem. 2013, 52, 7881–7888. [Google Scholar] [CrossRef]
- Hao, Y.; Murphy, G.L.; Bosbach, D.; Modolo, G.; Albrecht-Schmitt, T.E.; Alekseev, E.V. Porous Uranyl Borophosphates with Unique Three-Dimensional Open-Framework Structures. Inorg. Chem. 2017, 56, 9311–9320. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.C.; Hu, C.L.; Xu, X.; Kong, F.; Mao, J.G. SrGe2B2O8 and Sr3Ge2B6O16: Novel strontium borogermanates with three-dimensional and layered anionic architectures. Inorg. Chem. 2013, 52, 13644–13650. [Google Scholar] [CrossRef]
- Wu, S.; Beermann, O.; Wang, S.; Holzheid, A.; Depmeier, W.; Malcherek, T.; Modolo, G.; Alekseev, E.V.; Albrecht-Schmitt, T.E. Synthesis of uranium materials under extreme conditions: UO2[B3Al4O11(OH)], a complex 3D aluminoborate. Chem. Eur. J. 2012, 18, 4166–4169. [Google Scholar] [CrossRef]
- Klepov, V.V.; Juillerat, C.A.; Alekseev, E.V.; zur Loye, H.-C. Overstepping Löwenstein’s Rule—A Route to Unique Aluminophosphate Frameworks with Three-Dimensional Salt-Inclusion and Ion Exchange Properties. Inorg. Chem. 2019, 58, 724–736. [Google Scholar] [CrossRef]
- Mutailipu, M.; Poeppelmeier, K.R.; Pan, S. Borates: A Rich Source for Optical Materials. Chem. Rev. 2021, 121, 1130–1202. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, F.; Pan, S.; Yu, H.; Zhang, F.; Dong, X.; Han, S.; Dong, L.; Bai, C.; Wang, Z. Ba4(BO3)3(SiO4)·Ba3X (X = Cl, Br): New salt-inclusion borosilicate halides as potential deep UV nonlinear optical materials. J. Mater. Chem. C 2014, 2, 4257. [Google Scholar] [CrossRef]
- Miao, Z.; Yang, Y.; Wei, Z.; Yang, Z.; Yu, S.; Pan, S. NaCa5BO3(SiO4)2 with Interesting Isolated [BO3] and [SiO4] Units in Alkali- and Alkaline-Earth-Metal Borosilicates. Inorg. Chem. 2019, 58, 3937–3943. [Google Scholar] [CrossRef]
- Heyward, C.C.; McMillen, C.D.; Kolis, J.W. Hydrothermal Growth of Lanthanide Borosilicates: A Useful Approach to New Acentric Crystals Including a Derivative of Cappelenite. Inorg. Chem. 2014, 54, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, H.; Pan, S.; Huang, Z.; Yang, Z.; Su, X.; Poeppelmeier, K.R. Cs2B4SiO9: A Deep-Ultraviolet Nonlinear Optical Crystal. Angew. Chem. Int. Ed. 2013, 52, 3406–3410. [Google Scholar] [CrossRef] [PubMed]
- Heyward, C.; McMillen, C.D.; Kolis, J. Hydrothermal synthesis and structural analysis of new mixed oxyanion borates: Ba11B26O44(PO4)2(OH)6, Li9BaB15O27(CO3) and Ba3Si2B6O16. J. Solid State Chem. 2013, 203, 166–173. [Google Scholar] [CrossRef]
- Chang, L.-W.; Liu, H.-K.; Lii, K.-H. Flux synthesis and characterization of two barium hydroxyborosilicates with triple and single tetrahedral layers: Ba2[Si3B3O12(OH)] and Ba[Si2BO6(OH)]. J. Solid State Chem. 2020, 287, 121331. [Google Scholar] [CrossRef]
- Medvedev, A.G.; Mikhaylov, A.A.; Churakov, A.V.; Vener, M.V.; Tripol’skaya, T.A.; Cohen, S.; Lev, O.; Prikhodchenko, P.V. Potassium, Cesium, and Ammonium Peroxogermanates with Inorganic Hexanuclear Peroxo Bridged Germanium Anion Isolated from Aqueous Solution. Inorg. Chem. 2015, 54, 8058–8065. [Google Scholar] [CrossRef] [PubMed]
- Manceau, A.; Gorshkov, A.I.; Drits, V.A. Structural chemistry of Mn, Fe, Co, and Ni in manganese hydrous oxides: Part II. Information from EXAFS spectroscopy and electron and X-ray diffraction. Am. Mineral. 1992, 77, 1144–1157. [Google Scholar]
- Lee, C.-S.; Lin, C.-H.; Wang, S.-L.; Lii, K.-H. [Na7UIVO2(UVO)2(UV/VIO2)2Si4O16]: A mixed-valence uranium silicate. Angew. Chem. Int. Ed. 2010, 49, 4254–4256. [Google Scholar] [CrossRef]
- APEXIII Version 2016. 5-0, SAINT Version 7.60A, SADABS Version 2016/2; Bruker Analytical X-ray Systems: Madison, WI, USA, 2016.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Serezhkin, V.N. Multipurpose Crystallochemical Analysis with the Program Package TOPOS. IUCr CompComm Newslett. 2006, 7, 4–38. [Google Scholar]
- Alexandrov, E.V.; Blatov, V.A.; Kochetkov, A.V.; Proserpio, D.M. Underlying nets in three-periodic coordination polymers: Topology, taxonomy and prediction from a computer-aided analysis of the Cambridge Structural Database. CrystEngComm 2011, 13, 3947–3958. [Google Scholar] [CrossRef]
- Kropf, A.J.; Katsoudas, J.; Chattopadhyay, S.; Shibata, T.; Lang, E.A.; Zyryanov, V.N.; Ravel, B.; McIvor, K.; Kemner, K.M.; Scheckel, K.G.; et al. The New MRCAT (Sector 10) Bending Magnet Beamline at the Advanced Photon Source. AIP Conf. Proc. 2010, 1234, 299–302. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396–1399. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Rabenau, A. The role of hydrothermal synthesis in materials science. J. Mater. Educ. 1988, 10, 543–591. [Google Scholar]
- Stritzinger, J.T.; Alekseev, E.V.; Polinski, M.J.; Cross, J.N.; Eaton, T.M.; Albrecht-Schmitt, T.E. Further evidence for the stabilization of U(V) within a tetraoxo core. Inorg. Chem. 2014, 53, 5294–5299. [Google Scholar] [CrossRef]
- McMillen, C.D.; Kolis, J.W. Hydrothermal synthesis as a route to mineralogically-inspired structures. Dalton Trans. 2016, 45, 2772–2784. [Google Scholar] [CrossRef]
- Burns, P.C.; Grice, J.D.; Hawthorne, F.C. Borate Minerals. I. Polyhedral Clusters and Fundamental Building Blocks. Can. Mineral. 1995, 33, 1131–1151. [Google Scholar]
- Ewald, B.; Huang, Y.-X.; Kniep, R. Structural Chemistry of Borophosphates, Metalloborophosphates, and Related Compounds. Z. Anorg. Allg. Chem. 2007, 633, 1517–1540. [Google Scholar] [CrossRef]
- Qiu, J.; Ling, J.; Sieradzki, C.; Nguyen, K.; Wylie, E.M.; Szymanowski, J.E.S.; Burns, P.C. Expanding the Crystal Chemistry of Uranyl Peroxides: Four Hybrid Uranyl-Peroxide Structures Containing EDTA. Inorg. Chem. 2014, 53, 12084–12091. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Ling, J.; Jouffret, L.; Thomas, R.; Szymanowski, J.E.S.; Burns, P.C. Water-soluble multi-cage super tetrahedral uranyl peroxide phosphate clusters. Chem. Sci. 2014, 5, 303–310. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhadbhade, M.; Price, J.R.; Karatchevtseva, I.; Collison, D.; Lumpkin, G.R. Kinetics vs. thermodynamics: A unique crystal transformation from a uranyl peroxo-nanocluster to a nanoclustered uranyl polyborate. RSC Adv. 2014, 4, 34244–34247. [Google Scholar] [CrossRef]
- Thangavelu, S.G.; Cahill, C.L. Uranyl-Promoted Peroxide Generation: Synthesis and Characterization of Three Uranyl Peroxo [(UO2)2(O2)] Complexes. Inorg. Chem. 2015, 54, 4208–4221. [Google Scholar] [CrossRef]
- Qiu, J.; Dembowski, M.; Szymanowski, J.E.S.; Toh, W.C.; Burns, P.C. Time-Resolved X-ray Scattering and Raman Spectroscopic Studies of Formation of a Uranium-Vanadium-Phosphorus-Peroxide Cage Cluster. Inorg. Chem. 2016, 55, 7061–7067. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, M.; Colla, C.A.; Hickam, S.; Oliveri, A.F.; Szymanowski, J.E.S.; Oliver, A.G.; Casey, W.H.; Burns, P.C. Hierarchy of Pyrophosphate-Functionalized Uranyl Peroxide Nanocluster Synthesis. Inorg. Chem. 2017, 56, 5478–5487. [Google Scholar] [CrossRef]
- Qiu, J.; Spano, T.L.; Dembowski, M.; Kokot, A.M.; Szymanowski, J.E.; Burns, P.C. Sulfate-Centered Sodium-Icosahedron-Templated Uranyl Peroxide Phosphate Cages with Uranyl Bridged by μ-η1:η2 Peroxide. Inorg. Chem. 2017, 56, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.R. On the determination of uranium-oxygen bond lengths in dioxouranium(VI) compounds by Raman spectroscopy. J. Mol. Struct. 1989, 193, 295–300. [Google Scholar] [CrossRef]
- Frost, R.L.; Bouzaid, J.M.; Martens, W.N.; Reddy, B.J. Raman spectroscopy of the borosilicate mineral ferroaxinite. J. Raman Spectrosc. 2007, 38, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Bastians, S.; Crump, G.; Griffith, W.P.; Withnall, R. Raspite and studtite: Raman spectra of two unique minerals. J. Raman Spectrosc. 2004, 35, 726–731. [Google Scholar] [CrossRef]
- Dembowski, M.; Bernales, V.; Qiu, J.; Hickam, S.; Gaspar, G.; Gagliardi, L.; Burns, P.C. Computationally-Guided Assignment of Unexpected Signals in the Raman Spectra of Uranyl Triperoxide Complexes. Inorg. Chem. 2017, 56, 1574–1580. [Google Scholar] [CrossRef]
- McGrail, B.T.; Sigmon, G.E.; Jouffret, L.J.; Andrews, C.R.; Burns, P.C. Raman Spectroscopic and ESI-MS Characterization of Uranyl Peroxide Cage Clusters. Inorg. Chem. 2014, 53, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.L.; Čejka, J.; Weier, M.L.; Martens, W. Molecular structure of the uranyl silicates—A Raman spectroscopic study. J. Raman Spectrosc. 2006, 37, 538–551. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.L.; Čejka, J.; Weier, M.L.; Martens, W.; Kloprogge, J.T. A Raman and infrared spectroscopic study of the uranyl silicates-weeksite, soddyite and haiweeite. Spectrochim. Acta A 2006, 64, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Faulques, E.; Kalashnyk, N.; Massuyeau, F.; Perry, D.L. Spectroscopic markers for uranium phosphates: A vibronic study. RSC Adv. 2015, 5, 71219–71227. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | K1.78Na1.22[(UO2)BSi4O12] |
|---|---|
| space group | Cmce |
| formula weight (g/mol) | 682.77 |
| temperature (K) | 301(2) |
| a (Å) | 15.5471(19) |
| b (Å) | 14.3403(17) |
| c (Å) | 11.7315(15) |
| volume (Å3) | 2615.5(6) |
| Z | 8 |
| density (g cm−3) | 3.468 |
| crystal dimensions (mm3) | 0.04 × 0.03 × 0.02 |
| absorption coefficient (mm−1) | 13.460 |
| data collection and refinement | |
| collected reflections | 72,412 |
| independent reflections | 2963 |
| Rint | 0.0341 |
| refined restraints/parameters | 1/120 |
| goodness-of-fit on F2 | 1.251 |
| final R indices [I > 2σ(I)] | R1 = 0.0356 |
| wR2 = 0.0678 | |
| final R indices (all data) | R1 = 0.0376 |
| wR2 = 0.0686 | |
| largest diff. peak and hole (e−/Å3) | 4.154 and −2.158 |
| U-O | Experimental | Calculated | ||
|---|---|---|---|---|
| Fully Occupied Peroxo | Half-Occupied Peroxo | No Peroxo | ||
| U(1)-O(1) | 1.83 | 2.15 | 2.16 | 1.87 |
| U(1)-O(2) | 1.81 | 1.86 | 1.88 | 1.86 |
| U(1)-Operoxo | 1.94 | 2.16 | 2.17 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pace, K.A.; Klepov, V.V.; Smith, M.D.; Williams, T.; Morrison, G.; Lauterbach, J.A.; Misture, S.T.; zur Loye, H.-C. Hydrothermal Synthesis and Structural Investigation of a Crystalline Uranyl Borosilicate. Inorganics 2021, 9, 25. https://doi.org/10.3390/inorganics9040025
Pace KA, Klepov VV, Smith MD, Williams T, Morrison G, Lauterbach JA, Misture ST, zur Loye H-C. Hydrothermal Synthesis and Structural Investigation of a Crystalline Uranyl Borosilicate. Inorganics. 2021; 9(4):25. https://doi.org/10.3390/inorganics9040025
Chicago/Turabian StylePace, Kristen A., Vladislav V. Klepov, Mark D. Smith, Travis Williams, Gregory Morrison, Jochen A. Lauterbach, Scott T. Misture, and Hans-Conrad zur Loye. 2021. "Hydrothermal Synthesis and Structural Investigation of a Crystalline Uranyl Borosilicate" Inorganics 9, no. 4: 25. https://doi.org/10.3390/inorganics9040025
APA StylePace, K. A., Klepov, V. V., Smith, M. D., Williams, T., Morrison, G., Lauterbach, J. A., Misture, S. T., & zur Loye, H. -C. (2021). Hydrothermal Synthesis and Structural Investigation of a Crystalline Uranyl Borosilicate. Inorganics, 9(4), 25. https://doi.org/10.3390/inorganics9040025