
Exploring the Role of IL-17A in Oral Dysbiosis-Associated Periodontitis and Its Correlation with Systemic Inflammatory Disease




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Oral Bacteria in Maintaining Periodontal Tissue Homeostasis
Effects of Oral Bacteria on Innate Defense System
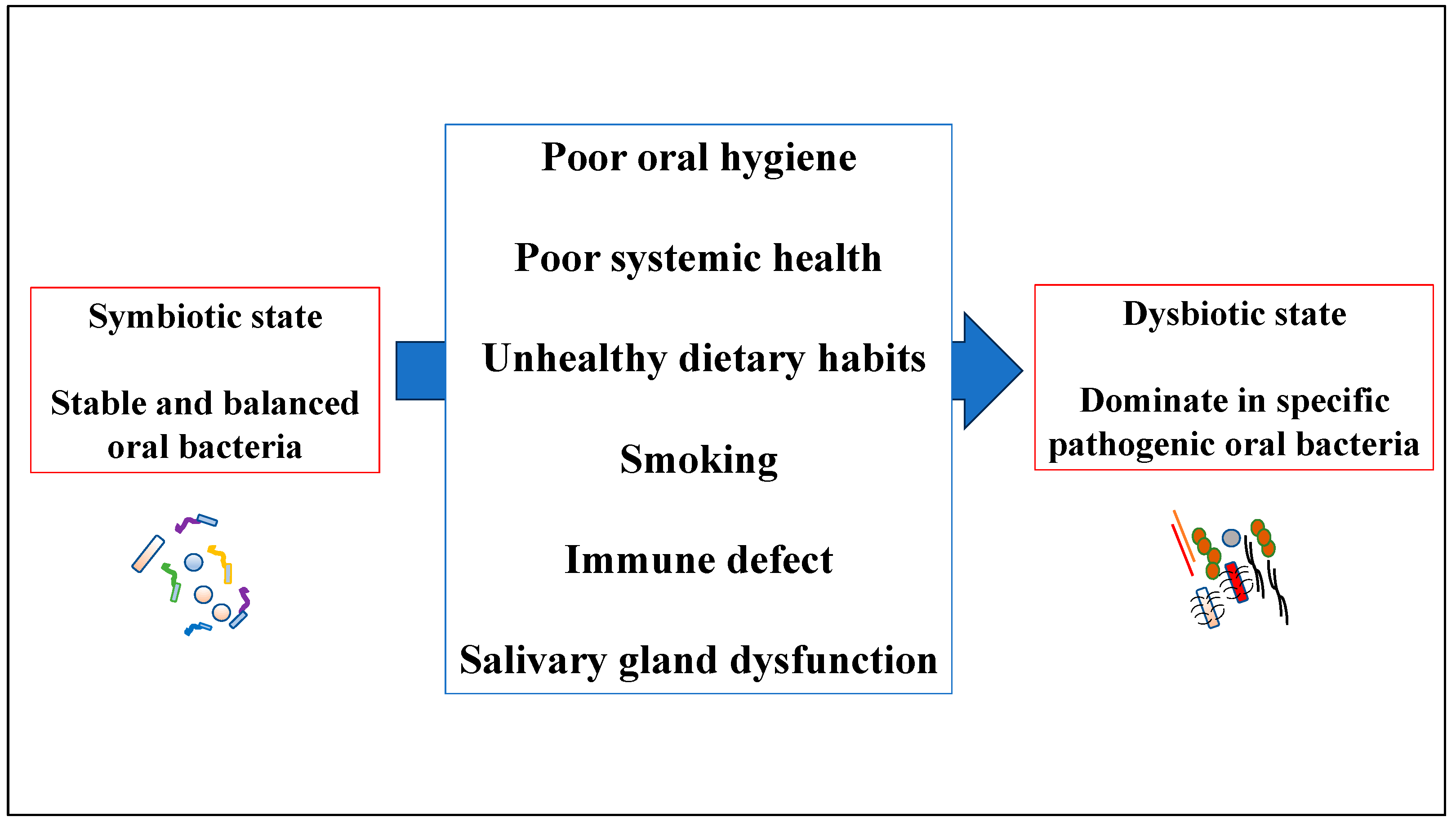
3. Overview of Human Oral Dysbiosis
3.1. Human Oral Dysbiosis in Periodontal Tissue
3.2. Interaction between Oral Dysbiosis and the Junctional Epithelium
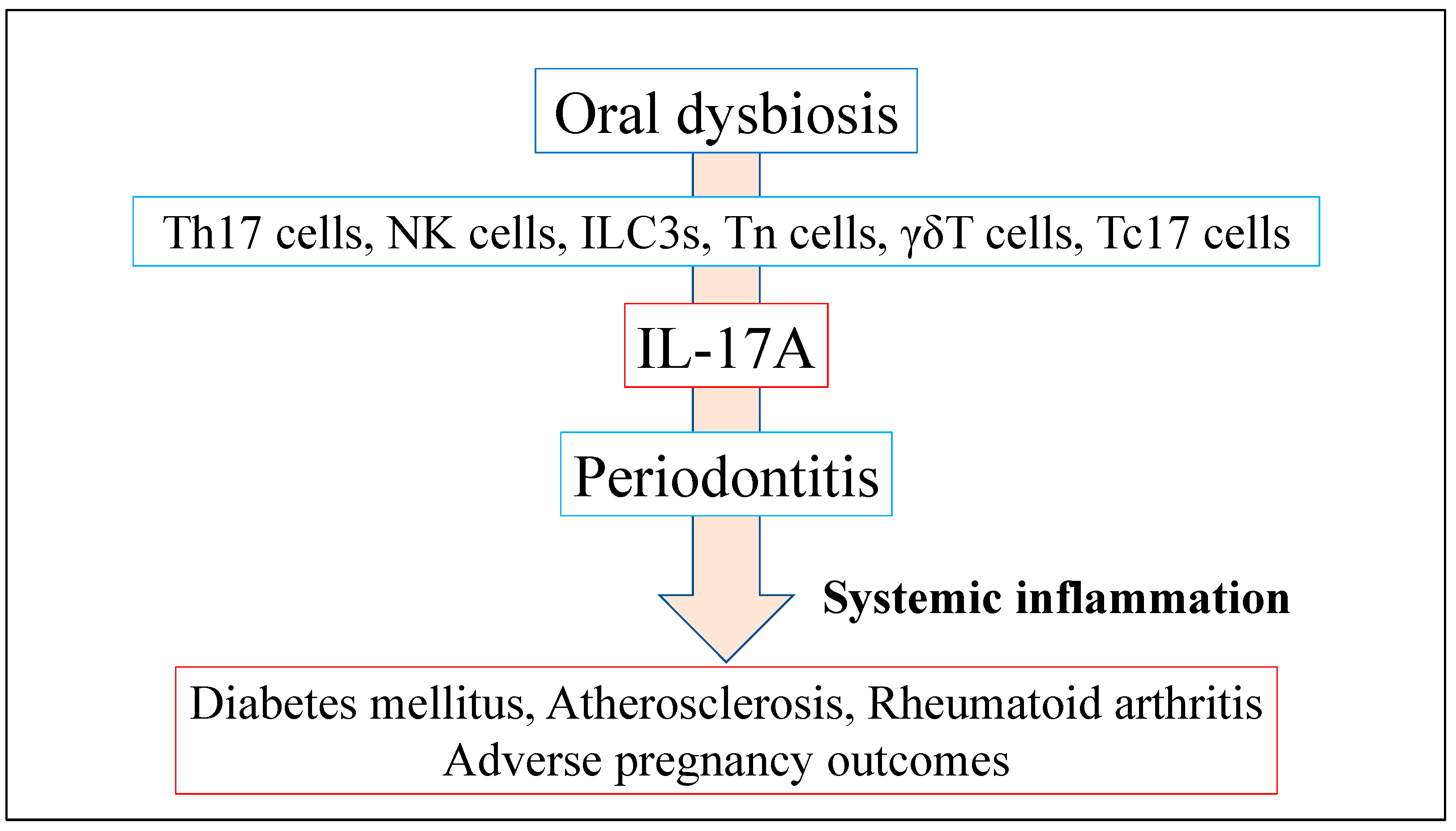
4. Oral Dysbiosis in Periodontitis and Systemic Disease
4.1. Role of IL-17A Production Induced by Oral Dysbiosis in Systemic Inflammatory Disease
4.2. Periodontitis, IL-17A, and Their Association with Diabetes Mellitus
4.3. Periodontitis, IL-17A, and Their Association with Atherosclerosis
4.4. Periodontitis, IL-17A, and Their Association with Rheumatoid Arthritis
4.5. Periodontitis, IL-17A, and Their Association with Adverse Pregnancy Outcomes
5. Prevention of Human Oral Dysbiosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Lin, W.; Jiang, W.; Hu, X.; Gao, L.; Ai, D.; Pan, H.; Niu, C.; Yuan, K.; Zhou, X.; Xu, C.; et al. Ecological Shifts of Supragingival Microbiota in Association with Pregnancy. Front. Cell Infect. Microbiol. 2018, 8, 24. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, K.; Chen, T.; Paster, B.J. A practical guide to the oral microbiome and its relation to health and disease. Oral. Dis. 2017, 23, 276–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belstrom, D.; Fiehn, N.E.; Nielsen, C.H.; Kirkby, N.; Twetman, S.; Klepac-Ceraj, V.; Paster, B.J.; Holmstrup, P. Differences in bacterial saliva profile between periodontitis patients and a control cohort. J. Clin. Periodontol. 2014, 41, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genom. Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [Green Version]
- Kolenbrander, P.E.; Andersen, R.N.; Blehert, D.S.; Egland, P.G.; Foster, J.S.; Palmer, R.J.J. Communication among oral bacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 486–505. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.D.; Slade, G.; Offenbacher, S. Oral disease, cardiovascular disease and systemic inflammation. Periodontology 2000 2000, 23, 110–120. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Palmer, R.J.J.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef]
- Marsh, P.D.; Moter, A.; Devine, D.A. Dental plaque biofilms: Communities, conflict and control. Periodontology 2000 2011, 55, 16–35. [Google Scholar] [CrossRef]
- Pérez-Chaparro, P.J.; Gonçalves, C.; Figueiredo, L.C.; Faveri, M.; Lobão, E.; Tamashiro, N.; Duarte, P.; Feres, M. Newly identified pathogens associated with periodontitis: A systematic review. J. Dent. Res. 2014, 93, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffen, A.L.; Beall, C.J.; Campbell, J.H.; Firestone, N.D.; Kumar, P.S.; Yang, Z.K.; Podar, M.; Leys, E.J. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012, 6, 1176–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstein, S.E.; Nelson, K.E.; Freire, M. Inflammatory Networks Linking Oral Microbiome with Systemic Health and Disease. J. Dent. Res. 2020, 99, 1131–1139. [Google Scholar] [CrossRef]
- La Rosa, G.R.M.; Gattuso, G.; Pedullà, E.; Rapisarda, E.; Nicolosi, D.; Salmeri, M. Association of oral dysbiosis with oral cancer development. Onco. Lett. 2020, 19, 3045–3058. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Abusleme, L.; Dupuy, A.K.; Dutzan, N.; Silva, N.; Burleson, J.A.; Strausbaugh, L.D.; Gamonal, J.; Diaz, P.I. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.A.; Diaz, P.I.; Van Dyke, T.E. The role of the microbiota in periodontal disease. Periodontology 2000 2020, 83, 14–25. [Google Scholar] [CrossRef]
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Sakallioğlu, E.E.; Lütfioğlu, M.; Sakallioğlu, U.; Diraman, E.; Keskiner, I. Fluid dynamics of gingiva in diabetic and systemically healthy periodontitis patients. Arch. Oral Biol. 2008, 53, 646–651. [Google Scholar] [CrossRef]
- Genco, R.J.; Graziani, F.; Hasturk, H. Effects of periodontal disease on glycemic control, complications, and incidence of diabetes mellitus. Periodontology 2000 2020, 83, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Scannapieco, F.A.; Bush, R.B.; Paju, S. Associations between periodontal disease and risk for atherosclerosis, cardiovascular disease, and stroke. A systematic review. Ann. Periodontol. 2003, 8, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, S.A.; Shirzaiy, M.; Risbaf, S. The Associations between Periodontitis and Respiratory Disease. J. Nepal Health Res. Counc. 2017, 15, 1–6. [Google Scholar] [CrossRef]
- Spiropoulou, A.; Zareifopoulos, N.; Bellou, A.; Spiropoulos, K.; Tsalikis, L. Review of the association between periodontitis and chronic obstructive pulmonary disease in smokers. Monaldi Arch. Chest Dis. 2019, 89, 83–89. [Google Scholar] [CrossRef]
- Suwannalai, P.; Trouw, L.A.; Toes, R.E.; Huizinga, T.W. Anti-citrullinated protein antibodies (ACPA) in early rheumatoid arthritis. Mod. Rheumatol. 2012, 22, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Bingham III, C.O.; Moni, M. Periodontal disease and rheumatoid arthritis: The evidence accumulates for complex pathobiologic interactions. Curr. Opin. Rheumatol. 2013, 25, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.W.; Fardini, Y.; Chen, C.; Iacampo, K.G.; Peraino, V.A.; Shamonki, J.M.; Redline, R.W. Term stillbirth caused by oral Fusobacterium nucleatum. Obstet. Gynecol. 2010, 115, 442–445. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.; York, B.R.; Michaud, D.S. Oral Health and Cancer. Curr. Oral Health Rep. 2019, 6, 130–137. [Google Scholar] [CrossRef]
- Shin, Y.J.; Choung, H.W.; Lee, J.H.; Rhyu, I.C.; Kim, H.D. Association of Periodontitis with Oral Cancer: A Case-Control Study. J. Dent. Res. 2019, 98, 526–533. [Google Scholar] [CrossRef]
- Mau, L.P.; Kuan, Y.C.; Tsai, Y.C.; Lin, J.J.; Huynh-Ba, G.; Weng, P.W.; Shieh, Y.S.; Cheng, W.C.; Huang, R.Y. Patients with chronic periodontitis present increased risk for osteoporosis: A population-based cohort study in Taiwan. J. Periodontal Res. 2017, 52, 922–929. [Google Scholar] [CrossRef]
- Penoni, D.C.; Vettore, M.V.; Torres, S.R.; Farias, M.L.F.; Leão, A.T.T. An investigation of the bidirectional link between osteoporosis and periodontitis. Arch. Osteoporos. 2019, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Awang, R.A.; Lappin, D.F.; MacPherson, A.; Riggio, M.; Robertson, D.; Hodge, P.; Ramage, G.; Culshaw, S.; Preshaw, P.M.; Taylor, J.; et al. Clinical associations between IL-17 family cytokines and periodontitis and potential differential roles for IL-17A and IL-17E in periodontal immunity. Inflamm. Res. 2014, 63, 1001–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.C.; van Asten, S.D.; Burns, L.A.; Evans, H.G.; Walter, G.J.; Hashim, A.; Hughes, F.J.; Taams, L.S. Periodontitis-associated pathogens P. gingivalis and A. actinomycetemcomitans activate human CD14(+) monocytes leading to enhanced Th17/IL-17 responses. Eur. J. Immunol. 2016, 46, 2211–2221. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Chen, Z.; Tu, S.Q.; Wei, J.M.; Hou, Y.L.; Kuang, Z.L.; Kang, X.N.; Ai, H. Role of Interleukin-17A in the Pathomechanisms of Periodontitis and Related Systemic Chronic Inflammatory Diseases. Front. Immunol. 2022, 13, 862415. [Google Scholar] [CrossRef]
- Figueredo, C.M.; Lira-Junior, R.; Love, R.M. T and B Cells in Periodontal Disease: New Functions in A Complex Scenario. Int. J. Mol. Sci. 2019, 20, 3349. [Google Scholar] [CrossRef] [Green Version]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef]
- Irie, K.; Tomofuji, T.; Ekuni, D.; Morita, M.; Shimazaki, Y.; Darveau, R.P. Impact of Oral Commensal Bacteria on Degradation of Periodontal Connective Tissue in Mice. J. Periodontol. 2015, 86, 899–905. [Google Scholar] [CrossRef]
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Irie, K.; Novince, C.M.; Darveau, R.P. Impact of the Oral Commensal Flora on Alveolar Bone Homeostasis. J. Dent. Res. 2014, 93, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Greer, A.; Irie, K.; Hashim, A.; Leroux, B.G.; Chang, A.M.; Curtis, M.A.; Darveau, R.P. Site-Specific Neutrophil Migration and CXCL2 Expression in Periodontal Tissue. J. Dent. Res. 2016, 95, 946–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meulen, T.A.; Harmsen, H.; Bootsma, H.; Spijkervet, F.; Kroese, F.; Vissink, A. The microbiome-systemic diseases connection. Oral Dis. 2016, 22, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Bäckhed, F. The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- Dahlen, G.; Fejerskov, O.; Manji, F. Current concepts and an alternative perspective on periodontal disease. BMC Oral Health 2020, 20, 235. [Google Scholar] [CrossRef]
- Zenobia, C.; Luo, X.L.; Hashim, A.; Abe, T.; Jin, L.; Chang, Y.; Jin, Z.C.; Sun, J.X.; Hajishengallis, G.; Curtis, M.A.; et al. Commensal bacteria-dependent select expression of CXCL2 contributes to periodontal tissue homeostasis. Cell Microbiol. 2013, 15, 1419–1426. [Google Scholar] [CrossRef] [Green Version]
- Skougaard, M.R. Cell renewal, with special reference to the gingival epithelium. Adv. Oral Biol. 1970, 4, 261–288. [Google Scholar]
- Fukuhara, D.; Irie, K.; Uchida, Y.; Kataoka, K.; Akiyama, K.; Ekuni, D.; Tomofuji, T.; Morita, M. Impact of commensal flora on periodontal immune response to lipopolysaccharide. J. Periodontol. 2018, 89, 1213–1220. [Google Scholar] [CrossRef]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Nguyen, T.; Brody, H.; Radaic, A.; Kapila, Y. Probiotics for periodontal health-Current molecular findings. Periodontology 2000 2021, 87, 254–267. [Google Scholar] [CrossRef]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the human oral microbiome during health and disease. mBio 2014, 5, e01012-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhakara, P.; Gupta, A.; Bhardwaj, A.; Wilson, A. Oral Dysbiotic Communities and Their Implications in Systemic Diseases. Dent. J. 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Marsh, P.D.; Devine, D.A. How is the development of dental biofilms influenced by the host? J. Clin. Periodontol. 2011, 38 (Suppl. 11), 28–35. [Google Scholar] [CrossRef]
- Hall, M.W.; Singh, N.; Ng, K.F.; Lam, D.K.; Goldberg, M.B.; Tenenbaum, H.C.; Neufeld, J.D.; Beiko, R.G.; Senadheera, D.B. Inter-personal diversity and temporal dynamics of dental, tongue, and salivary microbiota in the healthy oral cavity. NPJ Biofilms Microbiomes 2017, 3, 2. [Google Scholar] [CrossRef]
- Kilian, M.; Chapple, I.L.C.; Hannig, M.; Marsh, P.D.; Meuric, V.; Pedersen, A.M.L.; Tonetti, M.S.; Wade, W.G.; Zaura, E. The oral microbiome—An update for oral healthcare professionals. Br. Dent. J. 2016, 221, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Peters, B.A.; Dominianni, C.; Zhang, Y.; Pei, Z.; Yang, L.; Ma, Y.; Purdue, M.P.; Jacobs, E.J.; Gapstur, S.M.; et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016, 10, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D.; Head, D.A.; Devine, D.A. Ecological approaches to oral biofilms: Control without killing. Caries Res. 2015, 49 (Suppl. 1), 46–54. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.S.; Griffen, A.L.; Moeschberger, M.L.; Leys, E.J. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J. Clin. Microbiol. 2005, 43, 3944–3955. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Hasegawa, M.; Inohara, N. The Role of Oral Pathobionts in Dysbiosis during Periodontitis Development. J. Dent. Res. 2014, 93, 539–546. [Google Scholar] [CrossRef]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef] [PubMed]
- Tadjoedin, F.M.; Masulili, S.L.C.; Rizal, M.I.; Kusdhany, L.S.; Turana, Y.; Ismail, R.I.; Bachtiar, B.M. The Red and Orange Complex Subgingival Microbiome of Cognitive Impairment and Cognitively Normal Elderly with Periodontitis. Geriatrics 2022, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Rosier, B.T.; De Jager, M.; Zaura, E.; Krom, B.P. Historical and contemporary hypotheses on the development of oral diseases: Are we there yet? Front. Cell Infect. Microbiol. 2014, 4, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashishta, A.; Jimenez-Flores, E.; Klaes, C.K.; Tian, S.; Miralda, I.; Lamont, R.J.; Uriarte, S.M. Putative Periodontal Pathogens, Filifactor Alocis and Peptoanaerobacter Stomatis, Induce Differential Cytokine and Chemokine Production by Human Neutrophils. Pathogens 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Miralda, I.; Vashishta, A.; Rogers, M.N.; Lamont, R.J.; Uriarte, S.M. The emerging oral pathogen, Filifactor alocis, extends the functional lifespan of human neutrophils. Mol. Microbiol. 2022, 117, 1340–1351. [Google Scholar] [CrossRef]
- Jimenez Flores, E.; Tian, S.; Sizova, M.; Epstein, S.S.; Lamont, R.J.; Uriarte, S.M. Peptoanaerobacter stomatis Primes Human Neutrophils and Induces Granule Exocytosis. Infect. Immun. 2017, 85, e01043-16. [Google Scholar] [CrossRef] [Green Version]
- Fine, N.; Hassanpour, S.; Borenstein, A.; Sima, C.; Oveisi, M.; Scholey, J.; Cherney, D.; Glogauer, M. Distinct Oral Neutrophil Subsets Define Health and Periodontal Disease States. J. Dent. Res. 2016, 95, 931–938. [Google Scholar] [CrossRef]
- Lajqi, T.; Braun, M.; Kranig, S.A.; Frommhold, D.; Pöschl, J.; Hudalla, H. LPS Induces Opposing Memory-like Inflammatory Responses in Mouse Bone Marrow Neutrophils. Int. J. Mol. Sci. 2021, 22, 9803. [Google Scholar] [CrossRef]
- Gu, J.Y.; Liu, Y.J.; Zhu, X.Q.; Qiu, J.Y.; Sun, Y. Effects of Endotoxin Tolerance Induced by Porphyromonas gingivalis Lipopolysaccharide on Inflammatory Responses in Neutrophils. Inflammation 2020, 43, 1692–1706. [Google Scholar] [CrossRef]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontology 2000 2015, 69, 142–159. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.T.; Yao, X.T.; Peng, Q.; Chen, D.K. The protective and pathogenic roles of IL-17 in viral infections: Friend or foe? Open Biol. 2019, 9, 190109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Hassan, N.; Khatoon, K.; Mirza, M.A.; Naseef, P.P.; Kuruniyan, M.S.; Iqbal, Z. Periodontitis and Systemic Disorder-An Overview of Relation and Novel Treatment Modalities. Pharmaceutics 2021, 13, 1175. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, Y.; Xu, Y.; Sun, Y.; Li, L.; Chen, W. Effect of non-surgical periodontal therapy on the levels of Th17/Th1/Th2 cytokines and their transcription factors in Chinese chronic periodontitis patients. J. Clin. Periodontol. 2011, 38, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Abusleme, L.; Moutsopoulos, N.M. IL-17: Overview and role in oral immunity and microbiome. Oral Dis. 2017, 23, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Kajikawa, T.; Abusleme, L.; Greenwell-Wild, T.; Zuazo, C.E.; Ikeuchi, T.; Brenchley, L.; Abe, T.; Hurabielle, C.; Martin, D.; et al. A dysbiotic microbiome triggers T(H)17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 2018, 10, eaat0797. [Google Scholar] [CrossRef] [Green Version]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef] [Green Version]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.M.; Yoon, B.Y.; Her, Y.M.; Oh, H.J.; Lee, J.S.; Kim, K.W.; Lee, S.Y.; Woo, Y.J.; Park, K.S.; Park, S.H.; et al. IL-32 and IL-17 interact and have the potential to aggravate osteoclastogenesis in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kolls, J.K. Interluekin-17A (IL17A). Gene 2017, 614, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, M.K.; de Carvalho, A.C.G.; Alves, E.H.P.; da Silva, F.R.P.; Pessoa, L.D.S.; Vasconcelos, D.F.P. Genetic Factors and the Risk of Periodontitis Development: Findings from a Systematic Review Composed of 13 Studies of Meta-Analysis with 71,531 Participants. Int. J. Dent. 2017, 2017, 1914073. [Google Scholar] [CrossRef] [Green Version]
- Eskan, M.A.; Jotwani, R.; Abe, T.; Chmelar, J.; Lim, J.H.; Liang, S.; Ciero, P.A.; Krauss, J.L.; Li, F.; Rauner, M.; et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 2012, 13, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Xiao, E.; Mattos, M.; Vieira, G.H.A.; Chen, S.; Corrêa, J.D.; Wu, Y.; Albiero, M.L.; Bittinger, K.; Graves, D.T. Diabetes Enhances IL-17 Expression and Alters the Oral Microbiome to Increase Its Pathogenicity. Cell Host Microb. 2017, 22, 120–128.e4. [Google Scholar] [CrossRef] [Green Version]
- Chapple, I.L.; Genco, R. Diabetes and periodontal diseases: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Periodontol. 2013, 84 (Suppl. 4), S106–S112. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Xiao, E.; Graves, D.T. Diabetes mellitus related bone metabolism and periodontal disease. Int. J. Oral Sci. 2015, 7, 63–72. [Google Scholar] [CrossRef]
- Lalla, E.; Papapanou, P.N. Diabetes mellitus and periodontitis: A tale of two common interrelated diseases. Nat. Rev. Endocrinol. 2011, 7, 738–748. [Google Scholar] [CrossRef]
- Sanz, M.; Ceriello, A.; Buysschaert, M.; Chapple, I.; Demmer, R.T.; Graziani, F.; Herrera, D.; Jepsen, S.; Lione, L.; Madianos, P.; et al. Scientific evidence on the links between periodontal diseases and diabetes: Consensus report and guidelines of the joint workshop on periodontal diseases and diabetes by the International Diabetes Federation and the European Federation of Periodontology. J. Clin. Periodontol. 2018, 45, 138–149. [Google Scholar] [CrossRef]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef]
- Chen, B.; Wang, Z.; Wang, J.; Su, X.; Yang, J.; Zhang, Q.; Zhang, L. The oral microbiome profile and biomarker in Chinese type 2 diabetes mellitus patients. Endocrine 2020, 68, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Lux, R.; Klokkevold, P.; Chang, M.; Barnard, E.; Haake, S.; Li, H. The subgingival microbiome associated with periodontitis in type 2 diabetes mellitus. ISME J. 2020, 14, 519–530. [Google Scholar] [CrossRef]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, S.; Venugopal, R.; Chandrayan Rao, R.; Yuwanati, M.B.; Awasthi, H.; Jain, M. Association of chronic periodontitis and type 2 diabetes mellitus with salivary Del-1 and IL-17 levels. J. Oral Biol. Craniofac. Res. 2020, 10, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Mendis, S.; Davis, S.; Norrving, B. Organizational update: The world health organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke 2015, 46, e121–e122. [Google Scholar] [CrossRef]
- Schenkein, H.A.; Papapanou, P.N.; Genco, R.; Sanz, M. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontology 2000 2020, 83, 90–106. [Google Scholar] [CrossRef]
- Pavlic, V.; Peric, D.; Kalezic, I.S.; Madi, M.; Bhat, S.G.; Brkic, Z.; Staletovic, D. Identification of Periopathogens in Atheromatous Plaques Obtained from Carotid and Coronary Arteries. Biomed. Res. Int. 2021, 2021, 9986375. [Google Scholar] [CrossRef]
- Joo, J.Y.; Cha, G.S.; Chung, J.; Lee, J.Y.; Kim, S.J.; Choi, J. Peptide 19 of Porphyromonas gingivalis Heat Shock Protein Is a Potent Inducer of Low-Density Lipoprotein Oxidation. J. Periodontol. 2017, 88, e58–e64. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Nordlohne, J.; von Vietinghoff, S. Interleukin 17A in atherosclerosis—Regulation and pathophysiologic effector function. Cytokine 2019, 122, 154089. [Google Scholar] [CrossRef] [PubMed]
- Roszyk, E.; Puszczewicz, M. Role of human microbiome and selected bacterial infections in the pathogenesis of rheumatoid arthritis. Reumatologia 2017, 55, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Laugisch, O.; Wong, A.; Sroka, A.; Kantyka, T.; Koziel, J.; Neuhaus, K.; Sculean, A.; Venables, P.J.; Potempa, J.; Möller, B.; et al. Citrullination in the periodontium--a possible link between periodontitis and rheumatoid arthritis. Clin. Oral Investig. 2016, 20, 675–683. [Google Scholar] [CrossRef]
- Ayala-Herrera, J.L.; Abud-Mendoza, C.; Gonzalez-Amaro, R.F.; Espinosa-Cristobal, L.F.; Martinez-Martinez, R.E. Distribution of Porphyromonas gingivalis fimA genotypes in patients affected by rheumatoid arthritis and periodontitis. Acta Odontol. Scand. 2018, 76, 520–524. [Google Scholar] [CrossRef] [PubMed]
- de Molon, R.S.; Rossa, C.J.; Thurlings, R.M.; Cirelli, J.A.; Koenders, M.I. Linkage of Periodontitis and Rheumatoid Arthritis: Current Evidence and Potential Biological Interactions. Int. J. Mol. Sci. 2019, 20, 4541. [Google Scholar] [CrossRef] [Green Version]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef]
- Ceccarelli, F.; Saccucci, M.; Di Carlo, G.; Lucchetti, R.; Pilloni, A.; Pranno, N.; Luzzi, V.; Valesini, G.; Polimeni, A. Periodontitis and Rheumatoid Arthritis: The Same Inflammatory Mediators? Mediat. Inflamm. 2019, 2019, 6034546. [Google Scholar] [CrossRef]
- Bedoya, S.K.; Lam, B.; Lau, K.; Larkin III, J. Th17 cells in immunity and autoimmunity. Clin. Dev. Immunol. 2013, 2013, 986789. [Google Scholar] [CrossRef] [Green Version]
- Gümüş, P.; Buduneli, E.; Bıyıkoğlu, B.; Aksu, K.; Saraç, F.; Nile, C.; Lappin, D.; Buduneli, N. Gingival crevicular fluid, serum levels of receptor activator of nuclear factor-κB ligand, osteoprotegerin, and interleukin-17 in patients with rheumatoid arthritis and osteoporosis and with periodontal disease. J. Periodontol. 2013, 84, 1627–1637. [Google Scholar]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef]
- Madianos, P.N.; Bobetsis, Y.A.; Offenbacher, S. Adverse pregnancy outcomes (APOs) and periodontal disease: Pathogenic mechanisms. J. Clin. Periodontol. 2013, 40 (Suppl. 14), S170–S180. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hong, F.; Yu, X. Prevalence of periodontal disease in pregnancy: A systematic review and meta-analysis. J. Dent. 2022, 125, 104253. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, P.; Alessandri, R.; Eick, S.; Salvi, G.E.; Surbek, D.; Sculean, A. The potential association between gingival crevicular fluid inflammatory mediators and adverse pregnancy outcomes: A systematic review. Clin. Oral Investig. 2013, 17, 1453–1463. [Google Scholar] [CrossRef] [Green Version]
- Mohr, S.; Amylidi-Mohr, S.K.; Stadelmann, P.; Sculean, A.; Persson, R.; Eick, S.; Surbek, D.V. Systemic Inflammation in Pregnant Women With Periodontitis and Preterm Prelabor Rupture of Membranes: A Prospective Case-Control Study. Front. Immunol. 2019, 10, 2624. [Google Scholar] [CrossRef]
- Liang, S.; Ren, H.; Guo, H.; Xing, W.; Liu, C.; Ji, Y.; Jiang, H.; Zhang, P.; Du, M. Periodontal infection with Porphyromonas gingivalis induces preterm birth and lower birth weight in rats. Mol. Oral Microbiol. 2018, 33, 312–321. [Google Scholar] [CrossRef]
- Herrero, E.R.; Fernandes, S.; Verspecht, T.; Ugarte-Berzal, E.; Boon, N.; Proost, P.; Bernaerts, K.; Quirynen, M.; Teughels, W. Dysbiotic Biofilms Deregulate the Periodontal Inflammatory Response. J. Dent. Res. 2018, 97, 547–555. [Google Scholar] [CrossRef]
- Sälzer, S.; Graetz, C.; Dörfer, C.E.; Slot, D.E.; Van der Weijden, F.A. Contemporary practices for mechanical oral hygiene to prevent periodontal disease. Periodontology 2000 2020, 84, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Badersten, A.; Nilveus, R.; Egelberg, J. Effect of nonsurgical periodontal therapy. I. Moderately advanced periodontitis. J. Clin. Periodontol. 1981, 8, 57–72. [Google Scholar] [CrossRef]
- Leroy, R.; Bourgeois, J.; Verleye, L.; Toma, S. Should systemic antibiotics be prescribed in periodontal abscesses and pericoronitis? A systematic review of the literature. Eur. J. Oral Sci. 2022, 130, e12884. [Google Scholar] [CrossRef]
- Gorr, S.U.; Abdolhosseini, M. Antimicrobial peptides and periodontal disease. J. Clin. Periodontol. 2011, 38 (Suppl. 11), 126–141. [Google Scholar] [CrossRef]
- Bassetti, M.; Schär, D.; Wicki, B.; Eick, S.; Ramseier, C.A.; Arweiler, N.B.; Sculean, A.; Salvi, G.E. Anti-infective therapy of peri-implantitis with adjunctive local drug delivery or photodynamic therapy: 12-month outcomes of a randomized controlled clinical trial. Clin. Oral Implant. Res. 2014, 25, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.M.F.; Maltos, K.L.M.; Shehabeldin, M.S.; Thomas, L.L.; Zhuang, Z.; Yoshizawa, S.; Verdelis, K.; Gaffen, S.L.; Garlet, G.P.; Little, S.R.; et al. Local Sustained Delivery of Anti-IL-17A Antibodies Limits Inflammatory Bone Loss in Murine Experimental Periodontitis. J. Immunol. 2021, 206, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Weinberg, M.A.; Morris, A.; Reich, K. IL17A/F nanobody sonelokimab in patients with plaque psoriasis: A multicentre, randomised, placebo-controlled, phase 2b study. Lancet 2021, 397, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Moricke, R.; Dokoupilova, E.; Codding, C.; Neal, J.; Andersson, M.; Rohrer, S.; Richards, H. Secukinumab in Active Rheumatoid Arthritis: A Phase III Randomized, Double-Blind, Active Comparator- and Placebo-Controlled Study. Arthritis Rheumatol. 2017, 69, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irie, K.; Azuma, T.; Tomofuji, T.; Yamamoto, T. Exploring the Role of IL-17A in Oral Dysbiosis-Associated Periodontitis and Its Correlation with Systemic Inflammatory Disease. Dent. J. 2023, 11, 194. https://doi.org/10.3390/dj11080194
Irie K, Azuma T, Tomofuji T, Yamamoto T. Exploring the Role of IL-17A in Oral Dysbiosis-Associated Periodontitis and Its Correlation with Systemic Inflammatory Disease. Dentistry Journal. 2023; 11(8):194. https://doi.org/10.3390/dj11080194
Chicago/Turabian StyleIrie, Koichiro, Tetsuji Azuma, Takaaki Tomofuji, and Tatsuo Yamamoto. 2023. "Exploring the Role of IL-17A in Oral Dysbiosis-Associated Periodontitis and Its Correlation with Systemic Inflammatory Disease" Dentistry Journal 11, no. 8: 194. https://doi.org/10.3390/dj11080194
APA StyleIrie, K., Azuma, T., Tomofuji, T., & Yamamoto, T. (2023). Exploring the Role of IL-17A in Oral Dysbiosis-Associated Periodontitis and Its Correlation with Systemic Inflammatory Disease. Dentistry Journal, 11(8), 194. https://doi.org/10.3390/dj11080194