
Ginsenoside CK Inhibits Hypoxia-Induced Epithelial–Mesenchymal Transformation through the HIF-1α/NF-κB Feedback Pathway in Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines and Cell Culture
2.3. In Vitro Cytotoxicity Assay
2.4. Cell Colony Forming Assay
2.5. Flow Cytometry Analysis
2.6. Cell Migration and Invasion Assay
2.7. Immunofluorescence
2.8. RNA Isolation and RT-qPCR
2.9. Animal Studies of Hepatocellular Carcinoma
2.10. Western Blotting
2.11. Immunohistochemistry Assay
2.12. Statistical Analysis
3. Results
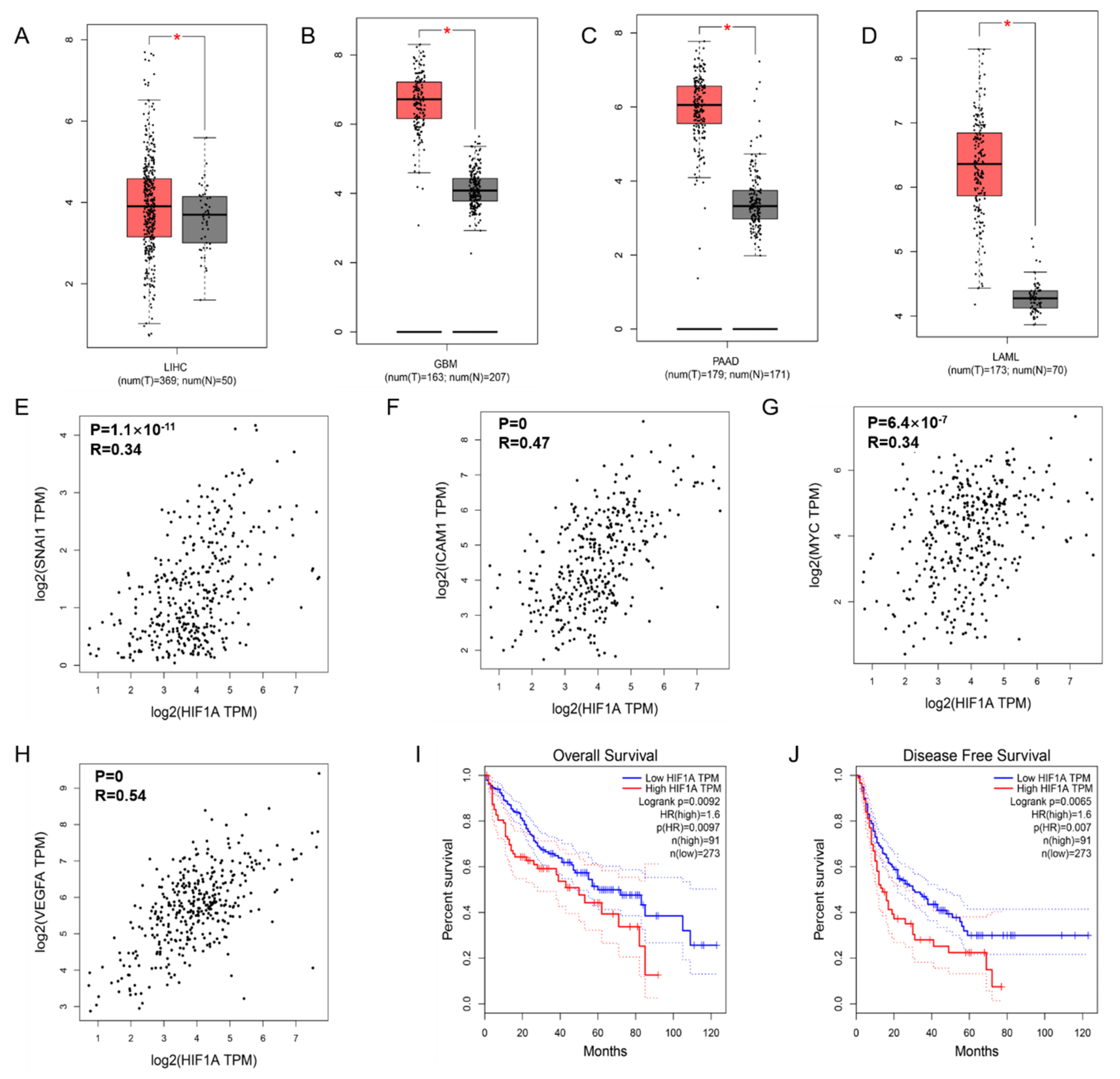
3.1. Transcription of HIF-1α in Cancers and the Correlation with NF-κB Signaling
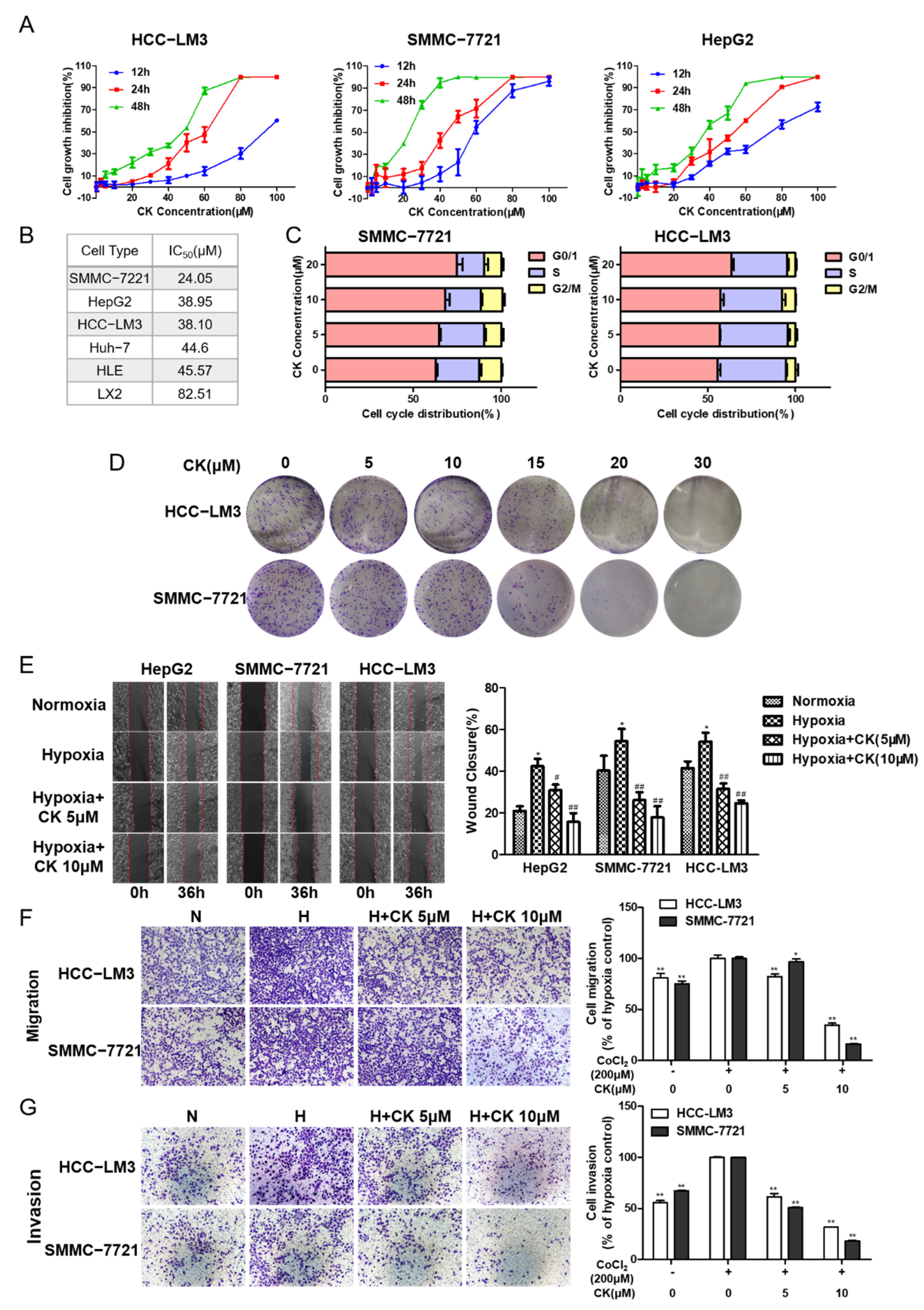
3.2. Ginsenoside CK Inhibits the Viability and Proliferation Characteristics of HCC Cells
3.3. Ginsenoside CK Suppresses Hypoxia-Induced Metastasis and Invasion of HCC Cells
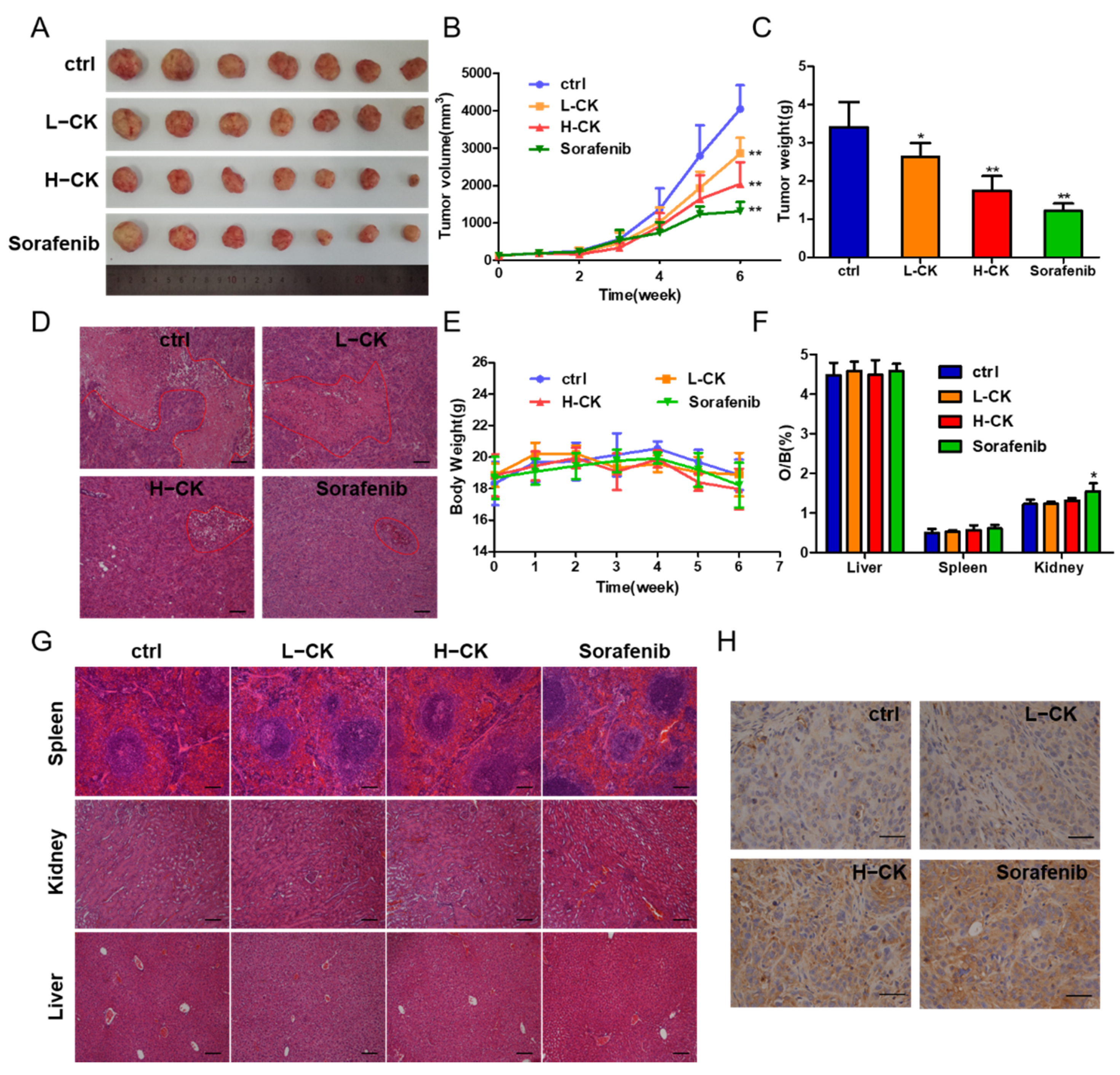
3.4. Ginsenoside CK Inhibits HCC Tumor Growth In Vivo
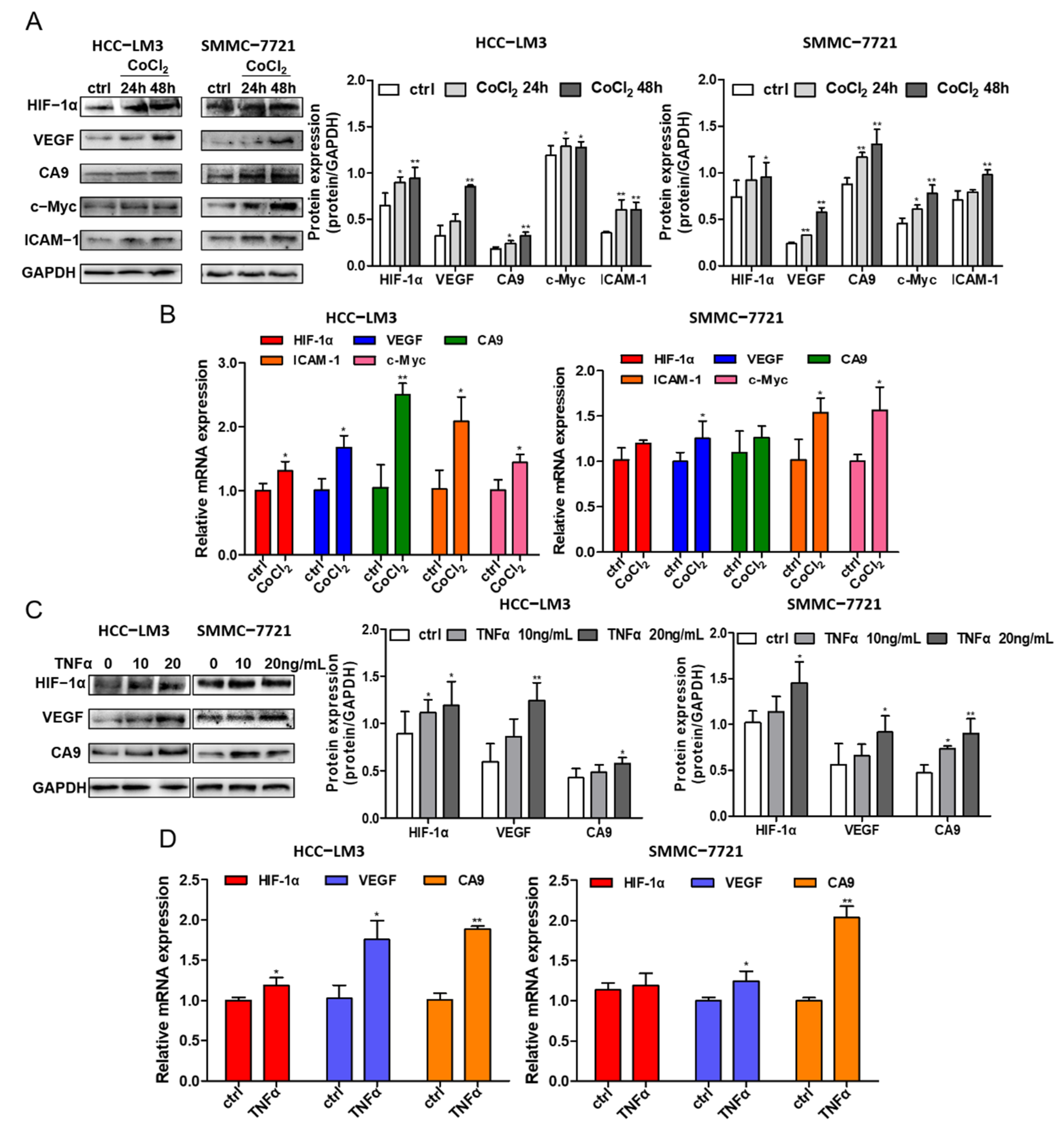
3.5. Crosstalk between HIF-1α and NF-κB Signaling in HCC Cells
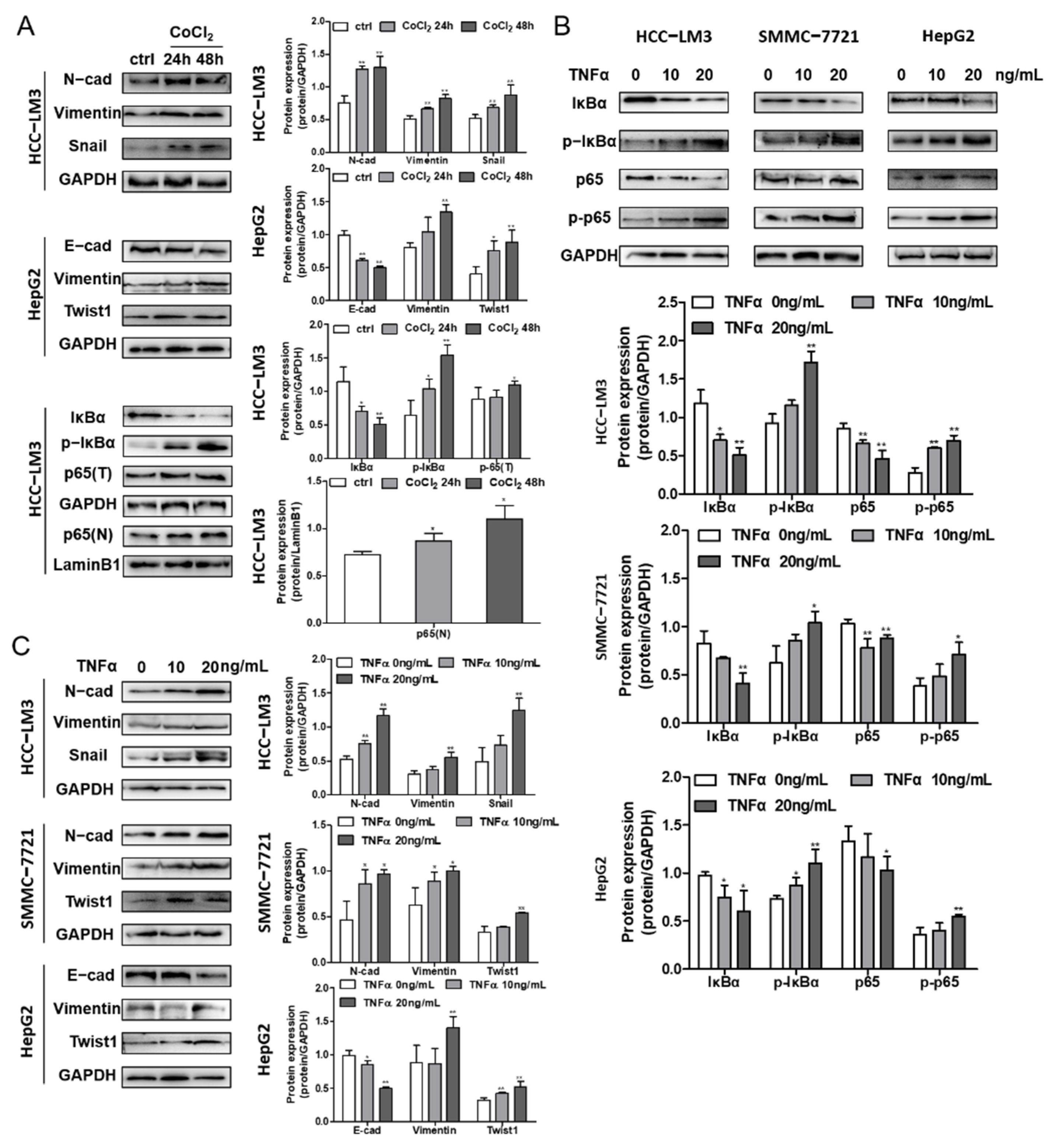
3.6. Hypoxia and NF-κB Signaling Stimulate EMT Progression in HCC Cells
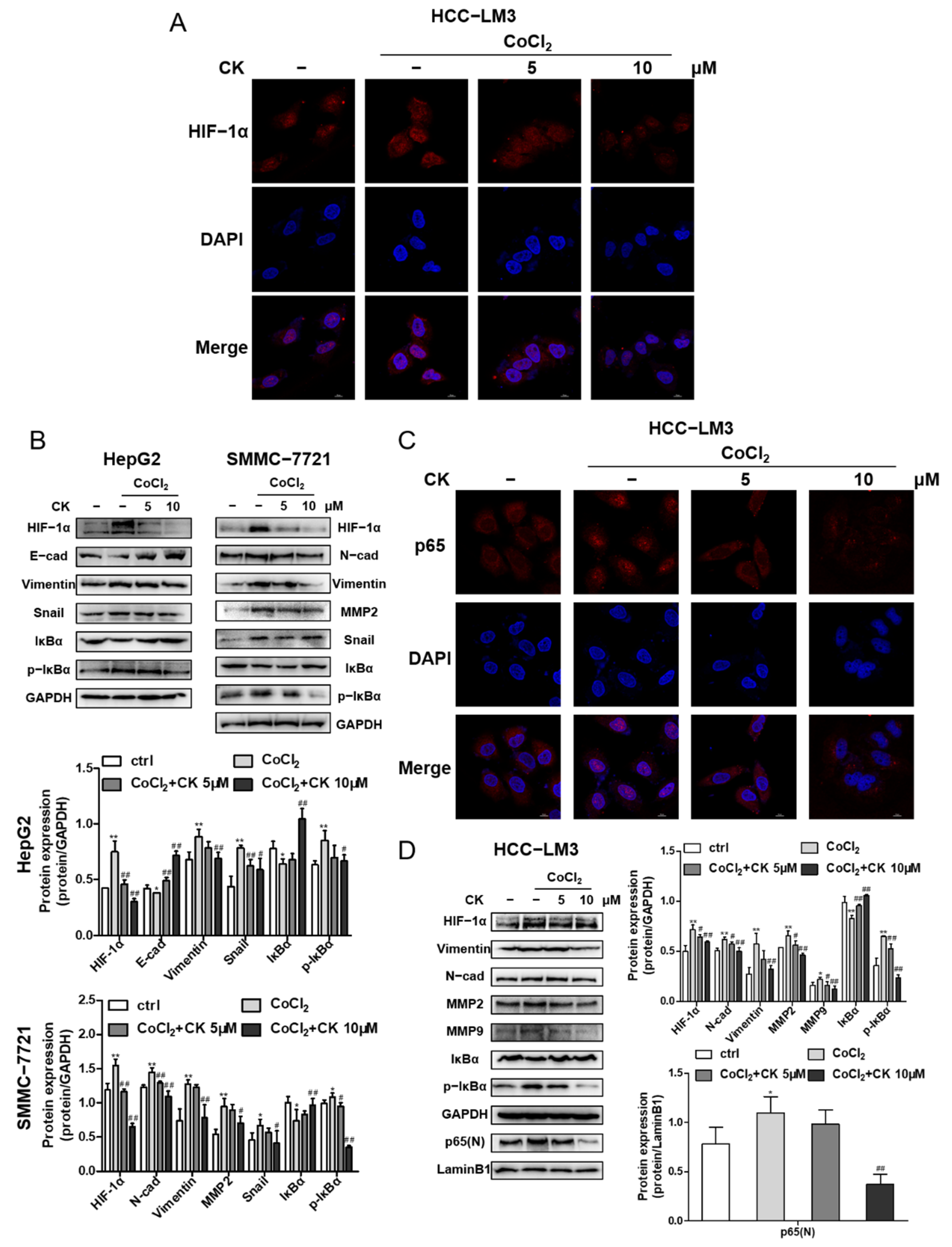
3.7. Ginsenoside CK Reversed Hypoxia-Induced EMT in HCC Cells
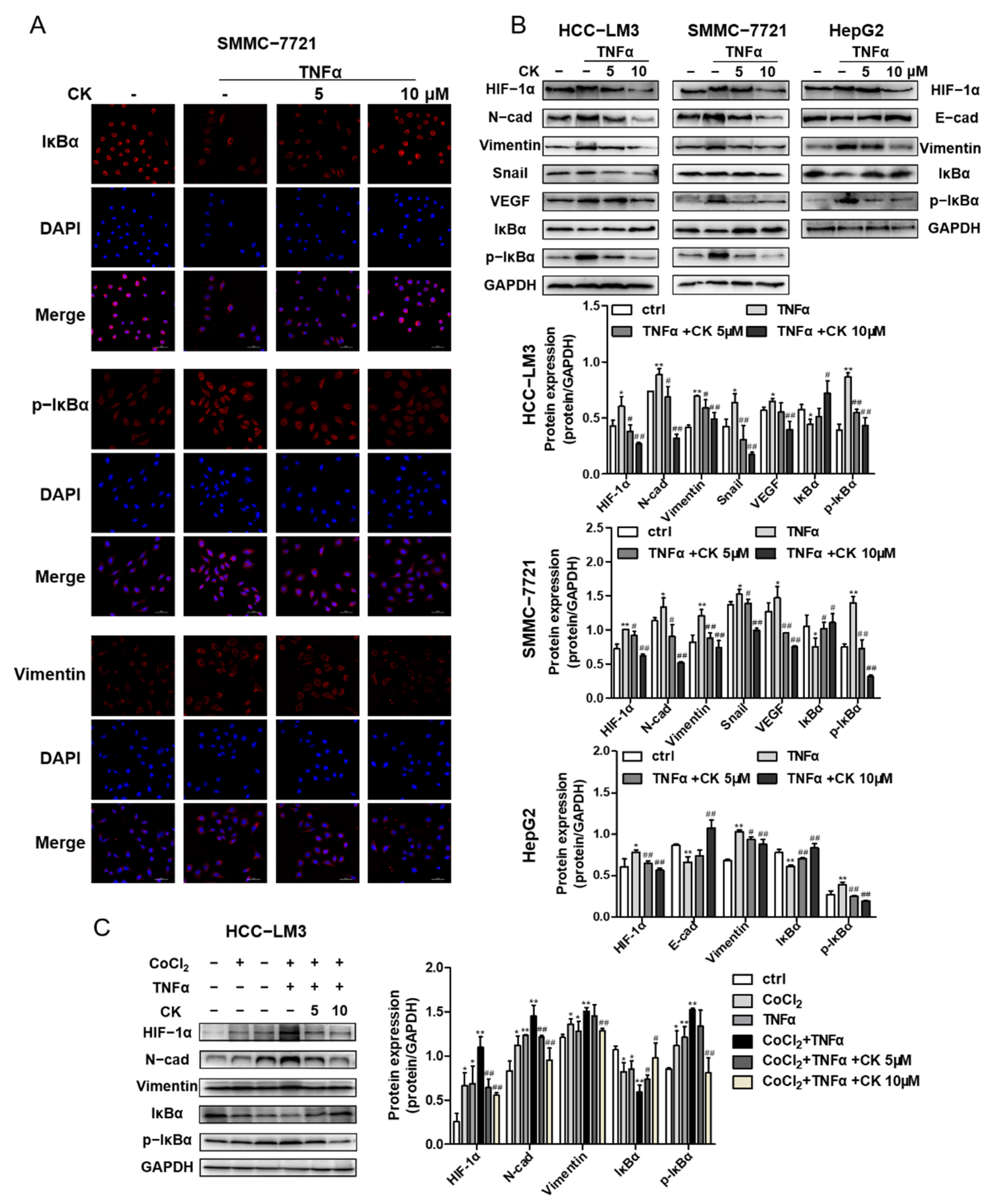
3.8. Ginsenoside CK Suppresses TNF-α-Induced EMT of HCC In Vitro
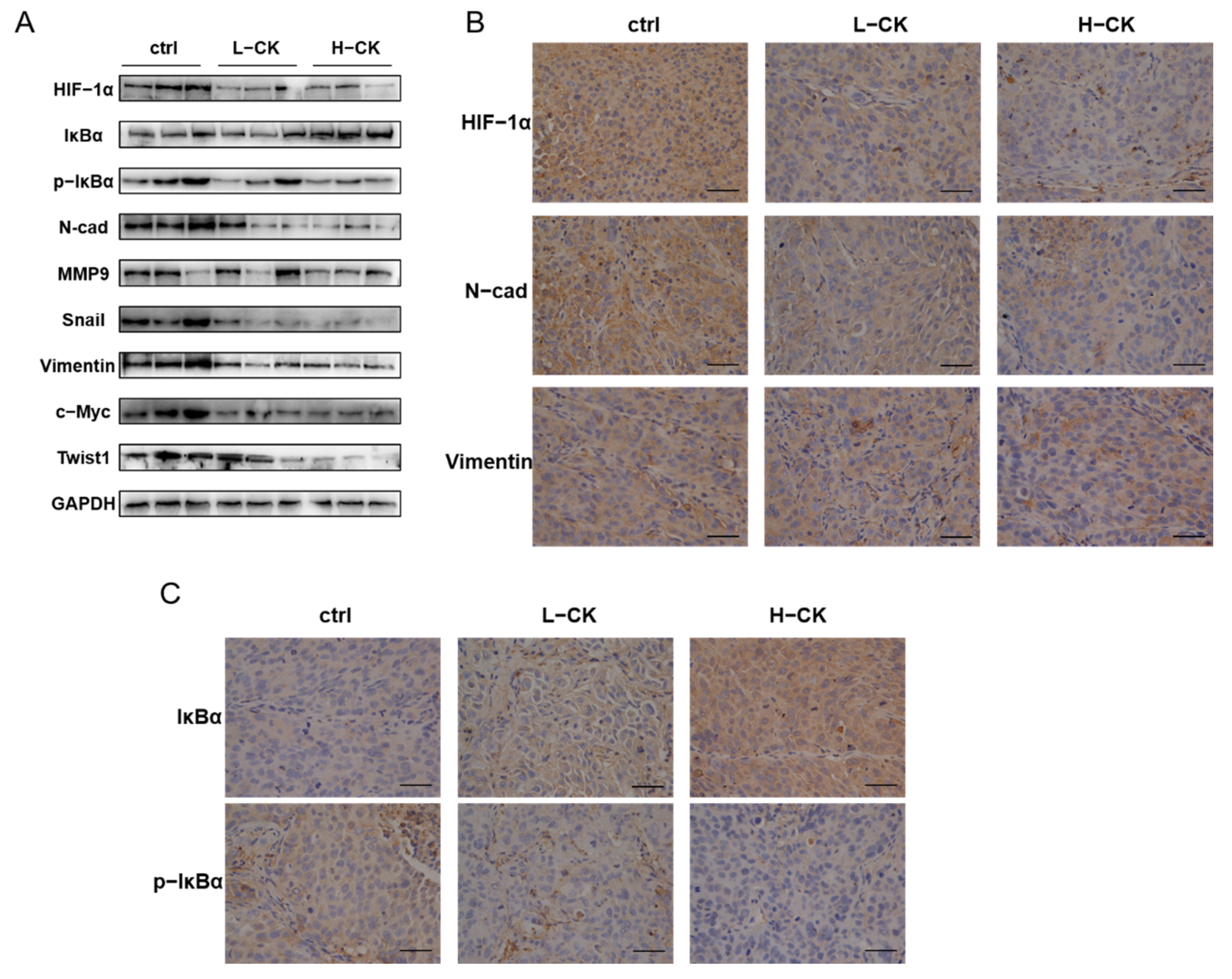
3.9. Ginsenoside CK Blocks the EMT of HCC In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Hseuabc, Y.C.; Lin, Y.C.; Rajendran, P.; Thigarajan, V.; Mathew, D.C.; Lin, K.Y.; Way, T.D.; Liao, J.W.; Yang, H.L. Antrodia salmonea suppresses invasion and metastasis in triple-negative breast cancer cells by reversing EMT through the NF-κB and Wnt/β-catenin signaling pathway. Food Chem. Toxicol. 2019, 124, 219–230. [Google Scholar]
- Wang, Q.; Cheng, Y.; Wang, Y.; Fan, Y.B.; Li, C.; Zhang, Y.; Wang, Y.D.; Dong, Q.; Ma, Y.J.; Teng, Y.E.; et al. Tamoxifen reverses epithelial–mesenchymal transition by demethylating miR-200c in triple-negative breast cancer cells. BMC Cancer 2017, 17, 492. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Harricharran, T.; Huaman, J.; Galuza, A.; Odumuwagun, O.; Tan, Y.; Ma, G.X.; Nguyen, M.T. Mechanisms of hepatocellular carcinoma progression. World J. Gastroenterol. 2019, 25, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Liu, D.; Ding, G.R.; Liao, P.F.; Zhang, J.W. Hypoxia-inducible factor-1α and Wnt/β-catenin signaling pathways promote the invasion of hypoxic gastric cancer cells. Mol. Med. Rep. 2015, 12, 3365–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giatromanolaki, A.; Harris, A.L. Tumour hypoxia, hypoxia signaling pathways and hypoxia inducible factor expression in human cancer. Anticancer Res. 2000, 21, 4317–4324. [Google Scholar]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragousis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.B.; Chen, T.X.; Mo, H.Y.; Chen, S.J.; Liu, Q.G.; Guo, C. Hypoxia-inducible long noncoding RNA NPSR1-AS1 promotes the proliferation and glycolysis of hepatocellular carcinoma cells by regulating the MAPK/ERK pathway. Biochem. Biophys. Res. Commun. 2020, 533, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Furuta, C.; Miyamoto, T.; Takagi, T.; Noguch, Y.; Kaneko, J.; Itoh, S.; Watanabe, T.; Itoh, F. Transforming growth factor-β signaling enhancement by long-term exposure to hypoxia in a tumor microenvironment composed of Lewis lung carcinoma cells. Cancer Sci. 2015, 106, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Chen, J.Y.; Lan, R.L.; Wang, Z.; Chen, R.Q. Hypoxia induced δ-Catenin to enhance mice hepatocellular carcinoma progression via Wnt signaling. Exp. Cell Res. 2019, 374, 94–103. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, G.; Li, X.W.; Zhang, Y.J.; Jiang, Y.; Shen, J.J.; Liu, J.; Wang, Q.L.; Zhu, J.; Feng, X.B.; et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1α in hepatocellular carcinoma. BMC Cancer 2013, 13, 108. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, Y.; Dou, C.; Xu, M.; Sun, L.; Wang, L.; Yao, B.; Li, Q.; Yang, W.; Tu, K.; et al. Hypoxia-induced up-regulation of VASP promotes invasiveness and metastasis of hepatocellular carcinoma. Theranostics 2018, 8, 4649–4663. [Google Scholar] [CrossRef]
- Pai, P.; Sukumar, S. HOX genes and the NF-κB pathway: A convergence of developmental biology, inflammation and cancer biology. Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1874, 188450. [Google Scholar] [CrossRef]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Hassanian, S.M. Role of the NF-κB signaling pathway in the pathogenesis of colorectal cancer. Gene 2019, 726, 144132. [Google Scholar] [CrossRef]
- Dai, W.; Wu, J.; Zhang, S.; Shi, B.; Xu, X.; Wang, D.; Wang, J. Genes directly regulated by NF-κB in human hepatocellular carcinoma HepG2. Int. J. Biochem. Cell Biol. 2017, 89, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bonello, S. The cross-talk between NF-κB and HIF-1: Further evidence for a significant liaison. Biochem. J. 2008, 412, 17–19. [Google Scholar] [CrossRef]
- Lin, C.W.; Wang, L.K.; Wang, S.P.; Chang, Y.L.; Wu, Y.Y.; Chen, H.Y.; Hsiao, T.H.; Lai, W.Y.; Lu, H.H.; Chang, Y.H.; et al. Daxx inhibits hypoxia-induced lung cancer cell metastasis by suppressing the HIF-1α/HDAC1/Slug axis. Nat. Commun. 2016, 7, 13867. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.J.; Chen, H.Y.; Ye, Z.; Deng, S.C.; Zhu, S.; Zeng, Z.; He, C.; Liu, M.L.; Huang, K.; Zhong, J.X.; et al. Hypoxia-induced LncRNA-BX111 promotes metastasis and progression of pancreatic cancer through regulating ZEB1 transcription. Oncogene 2018, 37, 5811–5828. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial–mesenchymal transition induced by TNF-α requires NF-κB-Mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.O.; Choi, E.; Shin, K.K.; Hong, Y.H.; Kim, H.G.; Jeong, D.; Hossain, M.A.; Kim, H.S.; Yi, Y.S.; Kim, D.; et al. Compound K, a ginsenoside metabolite, plays an anti-inflammatory role in macrophages by targeting the AKT1-mediated signaling pathway. J. Ginseng Res. 2018, 43, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Yang, Y.Y.; Ouyang, D.S.; Yang, G.P. A review of biotransformation and pharmacology of ginsenoside compound K. Fitoterapia 2015, 100, 208–220. [Google Scholar] [CrossRef]
- Chen, L.; Meng, Y.; Sun, Q.; Zhang, Z.Y.; Guo, X.Q.; Sheng, X.T.; Tai, G.H.; Cheng, H.R.; Zhou, Y.F. Ginsenoside compound K sensitizes human colon cancer cells to TRAIL-induced apoptosis via autophagy-dependent and -independent DR5 upregulation. Cell Death Dis. 2016, e2334. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, T.; Ma, C.Y.; Song, W.W.; Zhang, J.; Yu, Z.X. Ginsenoside metabolite compound K enhances the efficacy of cisplatin in lung cancer cells. J. Thoracic Dis. 2015, 7, 400–406. [Google Scholar]
- Zhang, K.; Li, Y. Effects of ginsenoside compound K combined with cisplatin on the proliferation, apoptosis and epithelial mesenchymal transition in MCF-7 cells of human breast cancer. Pharm. Biol. 2015, 54, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kwon, M.; Jang, J.P.; Sohng, J.K.; Jung, H.J. The ginsenoside metabolite compound K inhibits growth, migration and stemness of glioblastoma cells. Int. J. Oncol. 2017, 51, 414–424. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Zhu, H.Y.; Li, H.; Li, Y.; Zhao, B.; Jin, Y.H. Ginsenoside compound K inhibits NF-κB by targeting Annexin A2. J. Ginseng Res. 2018, 43, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Sung, J.H.; Lee, S.J.; Moon, C.K.; Lee, B.H. Antitumor activity of a novel ginseng saponin metabolite in human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer Lett. 1999, 144, 39–43. [Google Scholar] [CrossRef]
- Zhou, W.; Feng, M.Q.; Li, J.Y.; Zhou, P. Studies on the preparation, crystal structure and bioactivity of ginsenoside compound K. J. Asian Nat. Prod. Res. 2006, 8, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Ming, Y.L.; Song, G.; Chen, L.H.; Zheng, Z.Z.; Chen, Z.Y.; Ouyang, G.L.; Tong, Q.X. Anti-proliferation and apoptosis induced by a novel intestinal metabolite of ginseng saponin in human hepatocellular carcinoma cells. Cell Biol. Int. 2007, 31, 1265–1273. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [Green Version]
- Park, E.K.; Shin, Y.W.; Lee, H.U.; Kim, S.S.; Lee, Y.C.; Lee, B.Y.; Kim, D.H. Inhibitory effect of ginsenoside Rb1 and compound K on NO and prostaglandin E2 biosyntheses of RAW264.7 cells induced by lipopolysaccharide. Biol. Pharm. Bull. 2005, 28, 652–656. [Google Scholar] [CrossRef] [Green Version]
- Ming, Y.; Chen, Z.; Chen, L.; Lin, D.; Tong, Q.; Zheng, Z.; Song, G. Ginsenoside compound K attenuates metastatic growth of hepatocellular carcinoma, which is associated with the translocation of nuclear factor-κB p65 and reduction of matrix metalloproteinase-2/9. Planta Med. 2011, 77, 428–433. [Google Scholar] [CrossRef]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. BBA Mol. Cell Res. 2020, 1867, 118782. [Google Scholar]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Karin, M. NF-κB and cancer—Identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.X.; Sun, B.; Wang, S.J.; Gao, Y.; Zhang, Y.M.; Zhou, H.X.; Jia, G.; Wang, Y.W.; Kong, R.; Pan, S.H.; et al. Nuclear factor-κB-dependent epithelial to mesenchymal transition induced by HIF-1α activation in pancreatic cancer cells under hypoxic conditions. PLoS ONE 2011, 6, e23752. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Isaacs, J.S.; Lee, S.; Trepel, J.; Liu, Z.; Neckers, L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappaB activation. Biochem. J. 2003, 370, 1011–1017. [Google Scholar] [CrossRef]
- Tang, Z.F.; Li, C.W.; Kang, B.X.; Gao, G.; Li, C.; Zhang, Z.M. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.L.; Wang, T.; Wang, G.F.; Li, G.S.; Sun, C.F.; Jiang, Z.M.; Yang, J.R.; Li, Y.S.; You, Y.L.; Wu, X.R.; et al. Preclinical safety of ginsenoside compound K: Acute, and 26-week oral toxicity studies in mice and rats. Food Chem. Toxicol. 2019, 131, 110578. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Choi, M.S.; Jeung, W.; Ra, J.; Yoo, H.H.; Kim, D.H. Effects of gut microbiota on the pharmacokinetics of protopanaxadiol ginsenosides Rd, Rg3, F2, and compound K in healthy volunteers treated orally with red ginseng. J. Ginseng Res. 2020, 44, 611–618. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ma, X.; Fan, D. Ginsenoside CK Inhibits Hypoxia-Induced Epithelial–Mesenchymal Transformation through the HIF-1α/NF-κB Feedback Pathway in Hepatocellular Carcinoma. Foods 2021, 10, 1195. https://doi.org/10.3390/foods10061195
Zhang J, Ma X, Fan D. Ginsenoside CK Inhibits Hypoxia-Induced Epithelial–Mesenchymal Transformation through the HIF-1α/NF-κB Feedback Pathway in Hepatocellular Carcinoma. Foods. 2021; 10(6):1195. https://doi.org/10.3390/foods10061195
Chicago/Turabian StyleZhang, Jingjing, Xiaoxuan Ma, and Daidi Fan. 2021. "Ginsenoside CK Inhibits Hypoxia-Induced Epithelial–Mesenchymal Transformation through the HIF-1α/NF-κB Feedback Pathway in Hepatocellular Carcinoma" Foods 10, no. 6: 1195. https://doi.org/10.3390/foods10061195
APA StyleZhang, J., Ma, X., & Fan, D. (2021). Ginsenoside CK Inhibits Hypoxia-Induced Epithelial–Mesenchymal Transformation through the HIF-1α/NF-κB Feedback Pathway in Hepatocellular Carcinoma. Foods, 10(6), 1195. https://doi.org/10.3390/foods10061195