

Shrimp Lipid Prevents Endoplasmic Reticulum-Mediated Endothelial Cell Damage

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Shrimp Lipid (SL) Extraction
2.2. Fatty Acid Profiles
2.3. Cell Culture
2.4. Cell Viability Assay
2.5. Cytoprotective Activity for ER Stress Inducer
2.6. Nuclear Staining Assay
2.7. Western Blot Analysis
2.8. Immunofluorescence
2.9. Statistical Analysis
3. Results
3.1. Effects of SL Inducers on Human Endothelial EA.hy926 Cells
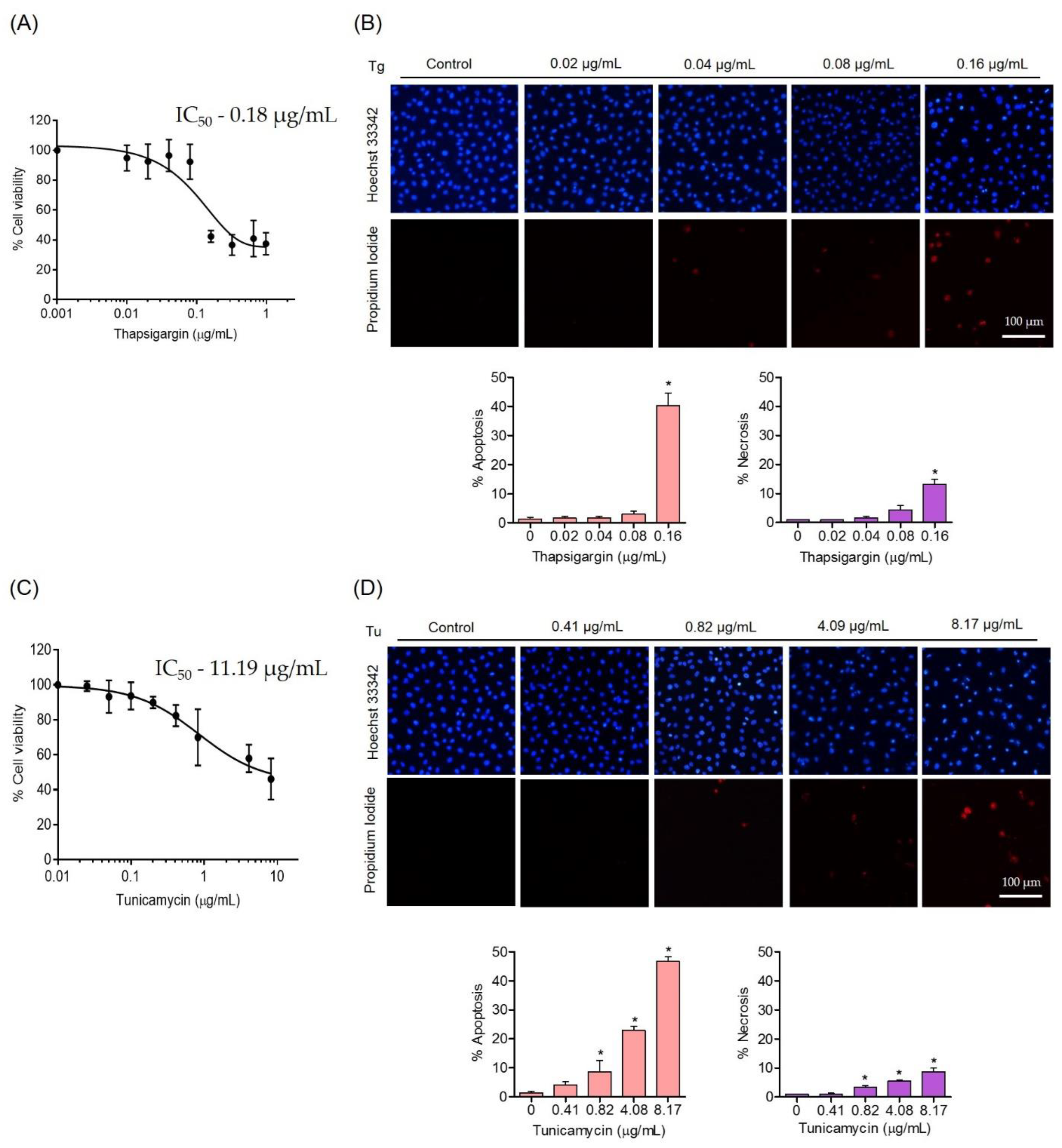
3.2. Effects of ER Stress Inducers on Human Endothelial Cells
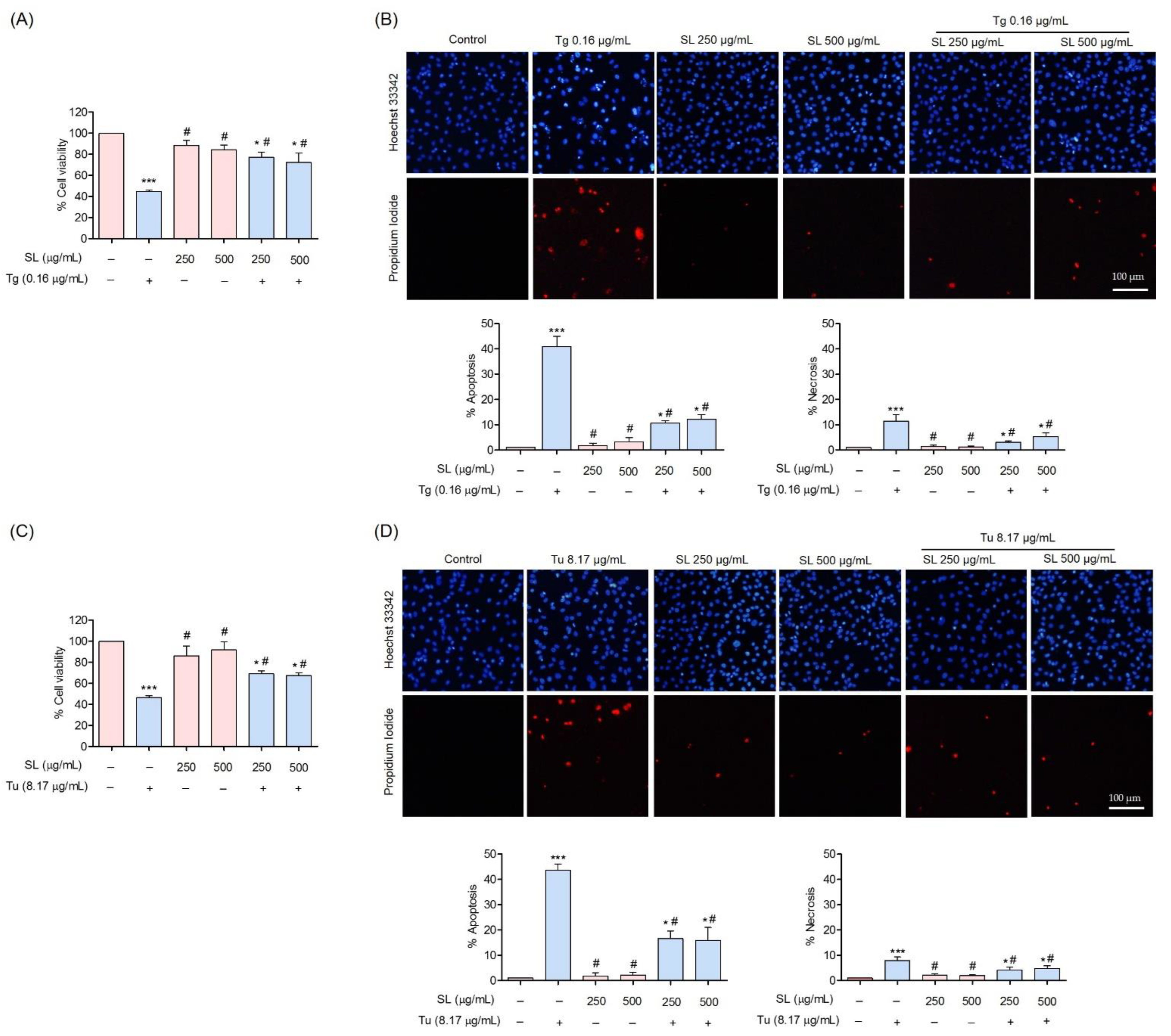
3.3. Protective Effect of SL on ER Stress-Induced Endothelial Cell Death
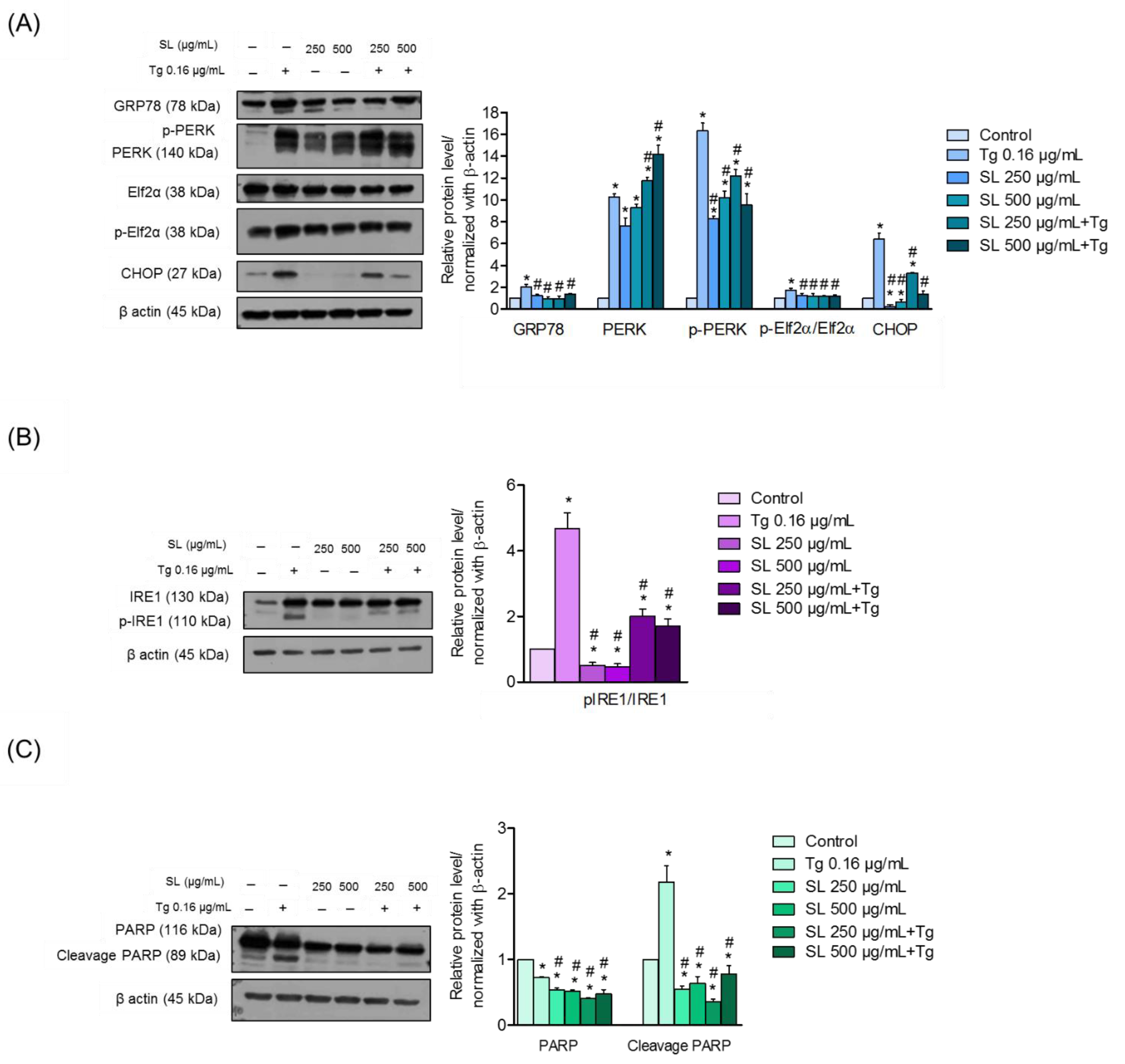
3.4. SL Prevents ER Stress Mediated Cell Death in Endothelial Cells via Reduction of ER Stress Response
3.5. SL Pretreatment Attenuated ER Stress-Mediated Apoptosis Induced by Thapsigargin
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- GLOBEFISH. Farmed Shrimp Stayed Stable in Asia, Increased Production in Latin America; GLOBEFISH: Rome, Italy, 2022. [Google Scholar]
- Cahú, T.B.; Santos, S.D.; Mendes, A.; Córdula, C.R.; Chavante, S.F.; Carvalho, L.B., Jr.; Nader, H.B.; Bezerra, R.S. Recovery of protein, chitin, carotenoids and glycosaminoglycans from Pacific white shrimp (Litopenaeus vannamei) processing waste. Process Biochem. 2012, 47, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Lordan, S.; Ross, R.P.; Stanton, C. Marine bioactives as functional food ingredients: Potential to reduce the incidence of chronic diseases. Mar. Drugs 2011, 9, 1056–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Xiong, Q.; Yin, Y.; Ling, Z.; Chen, S. The effects of fish oil on cardiovascular diseases: Systematical evaluation and recent advance. Front. Cardiovasc. Med. 2021, 8, 802306. [Google Scholar] [CrossRef] [PubMed]
- Kromhout, D.; Yasuda, S.; Geleijnse, J.M.; Shimokawa, H. Fish oil and omega-3 fatty acids in cardiovascular disease: Do they really work? Eur. Heart J. 2012, 33, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innes, J.K.; Calder, P.C. Marine Omega-3 (N-3) Fatty acids for cardiovascular health: An update for 2020. Int. J. Mol. Sci. 2020, 21, 1362. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Basha, B.; Samuel, S.M.; Triggle, C.R.; Ding, H. Endothelial dysfunction in diabetes mellitus: Possible involvement of endoplasmic reticulum stress? Exp. Diabetes Res. 2012, 2012, 481840. [Google Scholar] [CrossRef] [Green Version]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: A focus on the protective roles of heme oxygenase (HO)-1. Front.Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.C.; Weissman, A.M. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to ER stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Gulzar, S.; Benjakul, S. Ultrasound waves increase the yield and carotenoid content of lipid extracted from cephalothorax of Pacific white shrimp (Litopenaeus vannamei). Eur. J. Lipid Sci. Technol. 2018, 120, 1700495. [Google Scholar] [CrossRef]
- Raju, N.; Benjakul, S. Use of beta cyclodextrin to remove cholesterol and increase astaxanthin content in shrimp oil. Eur. J. Lipid Sci. Technol. 2020, 122, 1900242. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [Green Version]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef]
- Phadtare, I.; Vaidya, H.; Hawboldt, K.; Cheema, S.K. Shrimp oil extracted from shrimp processing by-product is a rich source of omega-3 fatty acids and astaxanthin-esters, and reveals potential anti-adipogenic effects in 3T3-L1 adipocytes. Mar. Drugs 2021, 19, 259. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory effect of astaxanthin on oxidative stress-induced mitochondrial dysfunction-a mini-review. Nutrients. 2018, 10, 1137. [Google Scholar] [CrossRef] [Green Version]
- Sztretye, M.; Dienes, B.; Gönczi, M.; Czirják, T.; Csernoch, L.; Dux, L.; Szentesi, P.; Keller-Pintér, A. Astaxanthin: A potential mitochondrial-targeted antioxidant treatment in diseases and with aging. Oxid. Med. Cell. Longev. 2019, 2019, 3849692. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Chyun, J.H.; Kim, Y.K.; Line, L.L.; Chew, B.P. Astaxanthin decreased oxidative stress and inflammation and enhanced immune response in humans. Nutr. Metab. 2010, 7, 18. [Google Scholar] [CrossRef]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef]
- Richard, D.; Kefi, K.; Barbe, U.; Bausero, P.; Visioli, F. Polyunsaturated fatty acids as antioxidants. Pharmacol. Res. 2008, 57, 451–455. [Google Scholar] [CrossRef]
- Oppedisano, F.; Macrì, R.; Gliozzi, M.; Musolino, V.; Carresi, C.; Maiuolo, J.; Bosco, F.; Nucera, S.; Caterina Zito, M.; Guarnieri, L.; et al. The anti-inflammatory and antioxidant properties of n-3 PUFAs: Their role in cardiovascular protection. Biomedicines. 2020, 8, 306. [Google Scholar] [CrossRef]
- Rodrigo, R.; Prieto, J.C.; Castillo, R. Cardioprotection against ischaemia/reperfusion by vitamins C and E plus n-3 fatty acids: Molecular mechanisms and potential clinical applications. Clin. Sci. 2013, 124, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Zhou, Y.; Wu, M.; Li, X.; Mai, K.; Ai, Q. ω-6 Polyunsaturated fatty acids (linoleic acid) activate both autophagy and antioxidation in a synergistic feedback loop via TOR-dependent and TOR-independent signaling pathways. Cell Death Dis. 2020, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Thayne, K.A.; Harris, M.; Shaikh, S.R.; Darden, T.M.; Lark, D.S.; Williams, J.M.; Chitwood, W.R.; Kypson, A.P.; Rodriguez, E. Do fish oil omega-3 fatty acids enhance antioxidant capacity and mitochondrial fatty acid oxidation in human atrial myocardium via PPARγ activation? Antioxid. Redox Signal. 2014, 21, 1156–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein quality control and endoplasmic reticulum stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [Green Version]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of apoptosis by the unfolded protein response. Methods Mol. Biol. 2009, 559, 191–204. [Google Scholar]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic reticulum stress: A critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef]
- Galán, M.; Kassan, M.; Kadowitz, P.J.; Trebak, M.; Belmadani, S.; Matrougui, K. Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Lenna, S.; Han, R.; Trojanowska, M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life 2014, 66, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Shang, Y.; Tao, J.; Zhang, J.; Sha, B. Endoplasmic reticulum stress signaling pathways: Activation and diseases. Curr. Protein. Pep. Sci. 2019, 20, 935–943. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Demay, Y.; Perochon, J.; Szuplewski, S.; Mignotte, B.; Gaumer, S. The PERK pathway independently triggers apoptosis and a Rac1/Slpr/JNK/Dilp8 signaling favoring tissue homeostasis in a chronic ER stress Drosophila model. Cell Death Dis. 2014, 5, e1452. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 2004, 71, 289–297. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty Acids | Amount (g/100 g Lipid) |
|---|---|
| C14:0 | 1.36 ± 0.01 |
| C15:0 | 0.79 ± 0.01 |
| C16:0 | 15.24 ± 0.05 |
| C17:0 | 1.39 ± 0.02 |
| C18:0 | 5.05 ± 0.06 |
| C20:0 | 2.30 ± 0.01 |
| C21:0 | 0.92 ± 0.01 |
| C23:0 | 2.44 ± 0.04 |
| C24:0 | 1.10 ± 0.02 |
| C16:1 | 0.52 ± 0.01 |
| C17:1 | 0.63 ± 0.03 |
| C18:1 n9t | 0.59 ± 0.01 |
| C18:1 n9c | 8.99 ± 0.02 |
| C20:1 | 0.56 ± 0.00 |
| C22:1 | 0.59 ± 0.01 |
| C24:1 | 0.77 ± 0.02 |
| C18:2 n6t | 0.66 ± 0.01 |
| C18:2 n3c | 8.09 ± 0.03 |
| C18:3 n3c | 0.61 ± 0.01 |
| C20:2 n6c | 1.33 ± 0.02 |
| C20:3 n6c | 0.54 ± 0.01 |
| C20:4 n6c | 0.56 ± 0.01 |
| C20:5 n3c | 5.66 ± 0.03 |
| C22:2 n6c | 0.57 ± 0.01 |
| C22:6 n3c | 7.11 ± 0.04 |
| SFA | 30.59 ± 0.02 |
| MUFA | 12.65 ± 0.01 |
| PUFA | 25.13 ± 0.01 |
| Total | 68.37 ± 0.02 |
| Astaxanthin content (mg/g lipid) | 1.08 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ei, Z.Z.; Benjakul, S.; Buamard, N.; Visuttijai, K.; Chanvorachote, P. Shrimp Lipid Prevents Endoplasmic Reticulum-Mediated Endothelial Cell Damage. Foods 2022, 11, 3076. https://doi.org/10.3390/foods11193076
Ei ZZ, Benjakul S, Buamard N, Visuttijai K, Chanvorachote P. Shrimp Lipid Prevents Endoplasmic Reticulum-Mediated Endothelial Cell Damage. Foods. 2022; 11(19):3076. https://doi.org/10.3390/foods11193076
Chicago/Turabian StyleEi, Zin Zin, Soottawat Benjakul, Natchaphol Buamard, Kittichate Visuttijai, and Pithi Chanvorachote. 2022. "Shrimp Lipid Prevents Endoplasmic Reticulum-Mediated Endothelial Cell Damage" Foods 11, no. 19: 3076. https://doi.org/10.3390/foods11193076
APA StyleEi, Z. Z., Benjakul, S., Buamard, N., Visuttijai, K., & Chanvorachote, P. (2022). Shrimp Lipid Prevents Endoplasmic Reticulum-Mediated Endothelial Cell Damage. Foods, 11(19), 3076. https://doi.org/10.3390/foods11193076