


The Detection of Foodborne Pathogenic Bacteria in Seafood Using a Multiplex Polymerase Chain Reaction System
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Primers
2.3. DNA Extraction and Concentration Measurement
2.4. Optimization of PCR Reaction Conditions
2.5. Sensitivity of Single PCR
2.6. Optimization of Multiplex Primer Ratios and Detection of Sensitivity of Multiplex PCR
2.7. Specificity of Multiplex PCR and Stability of Multiplex PCR
2.8. Evaluation of Artificially Contaminated Samples
3. Results
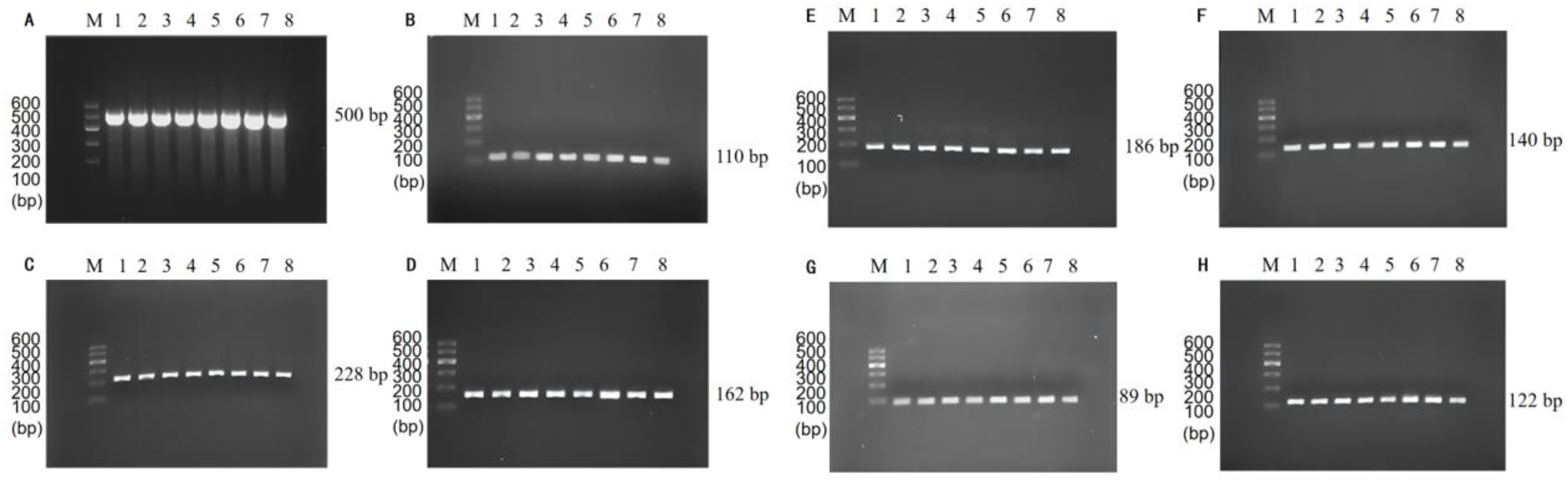
3.1. Optimization of PCR Annealing Temperature
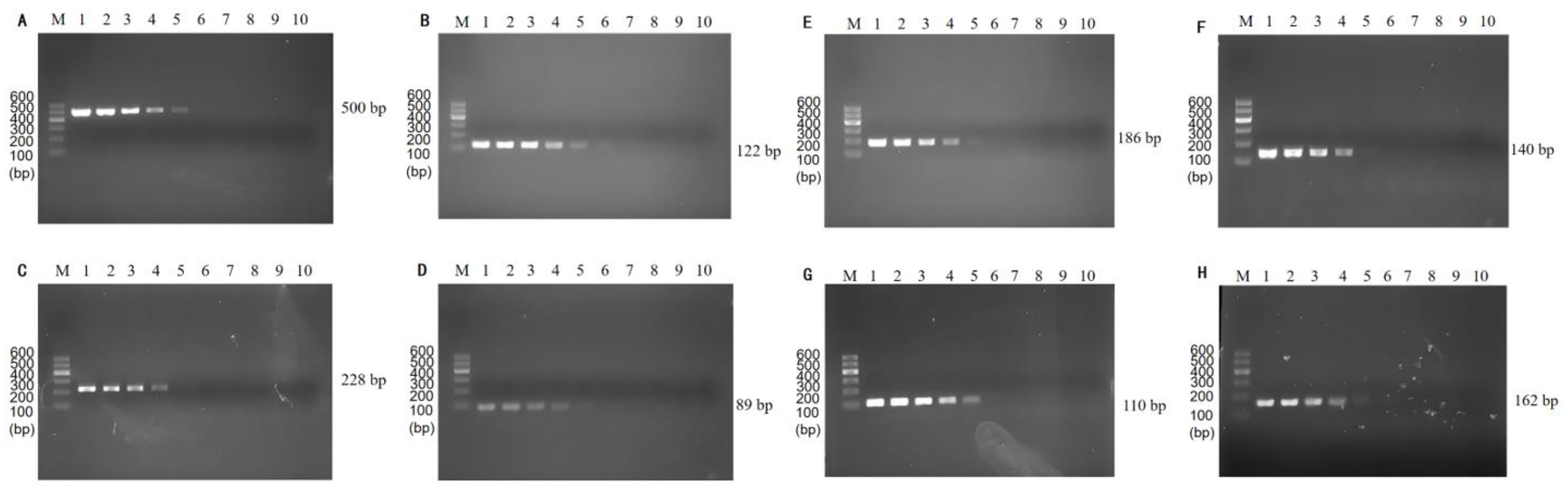
3.2. Single PCR Assay Sensitivity Validation and Traditional Method
3.3. Optimization of Multiplex PCR Primer Ratios and Sensitivity
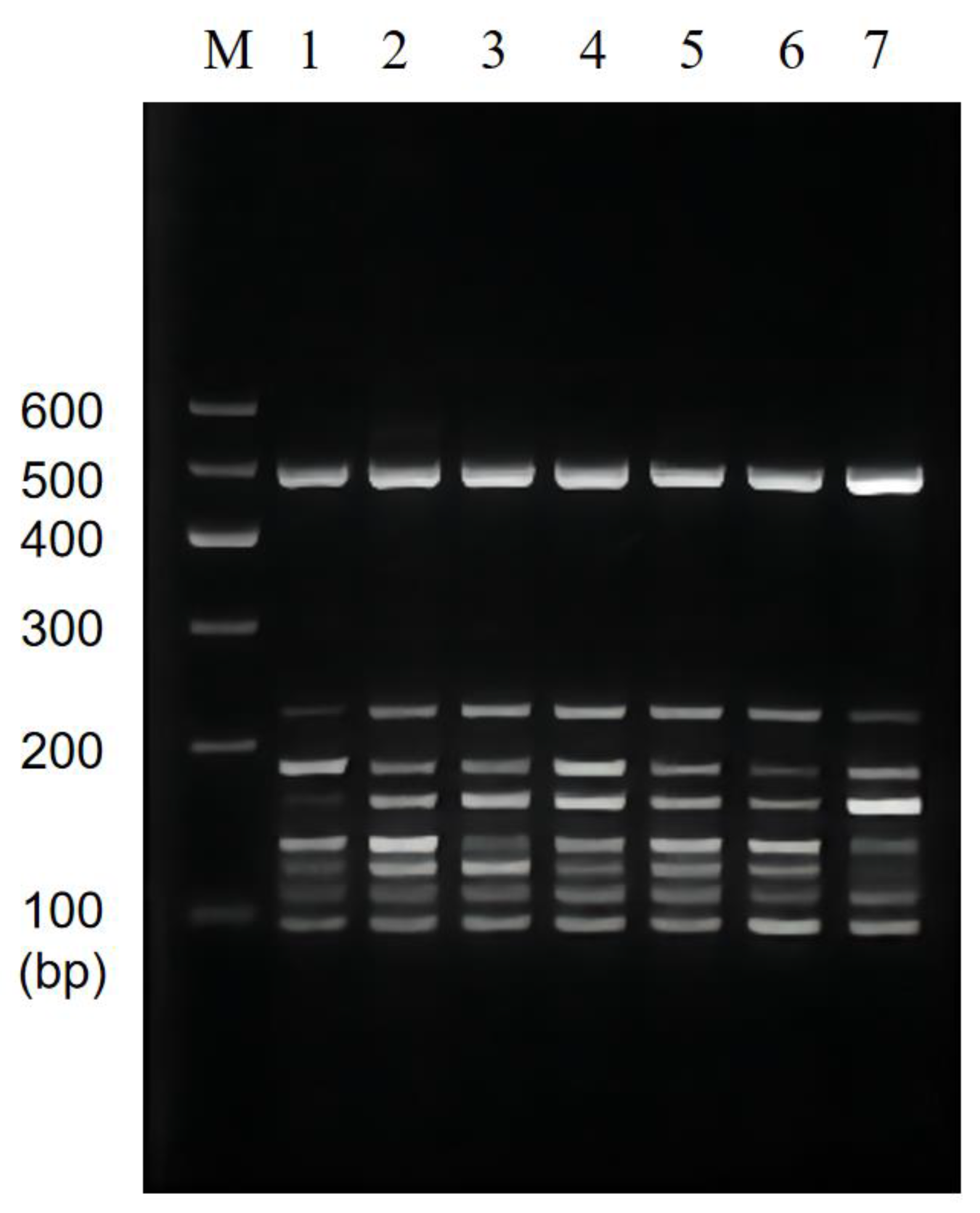
3.4. Results of Multiplex PCR Specificity and Stability
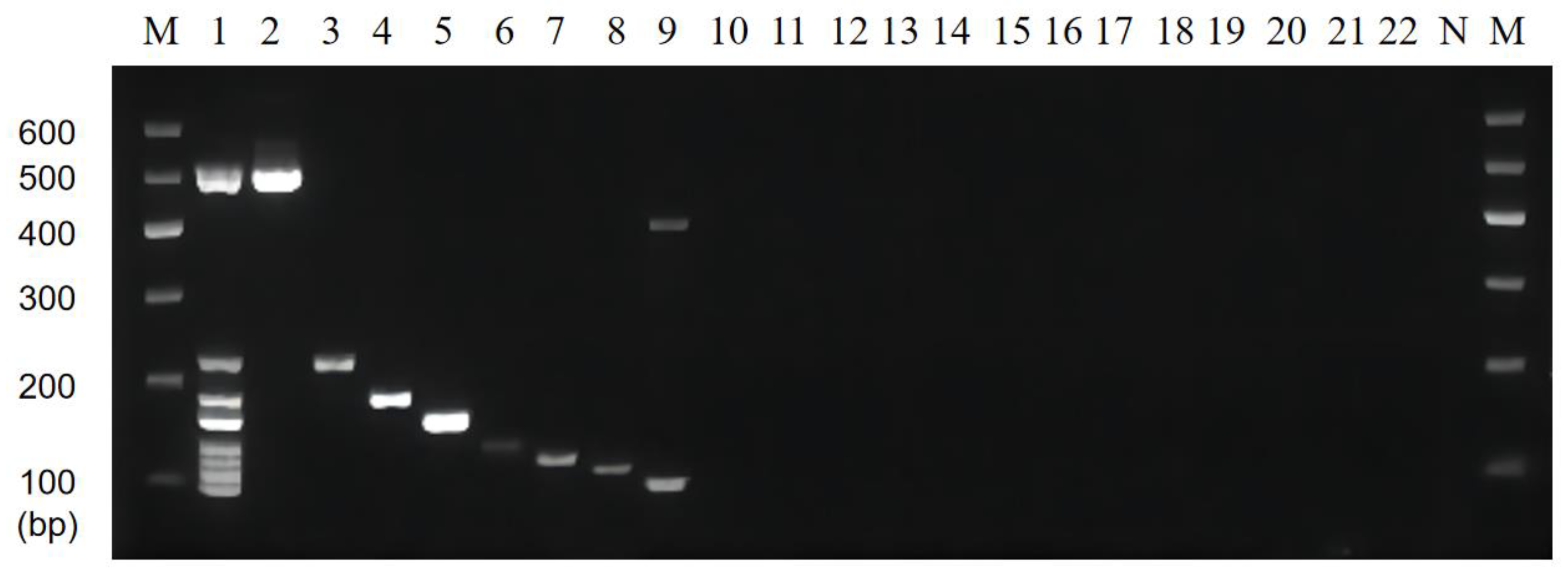
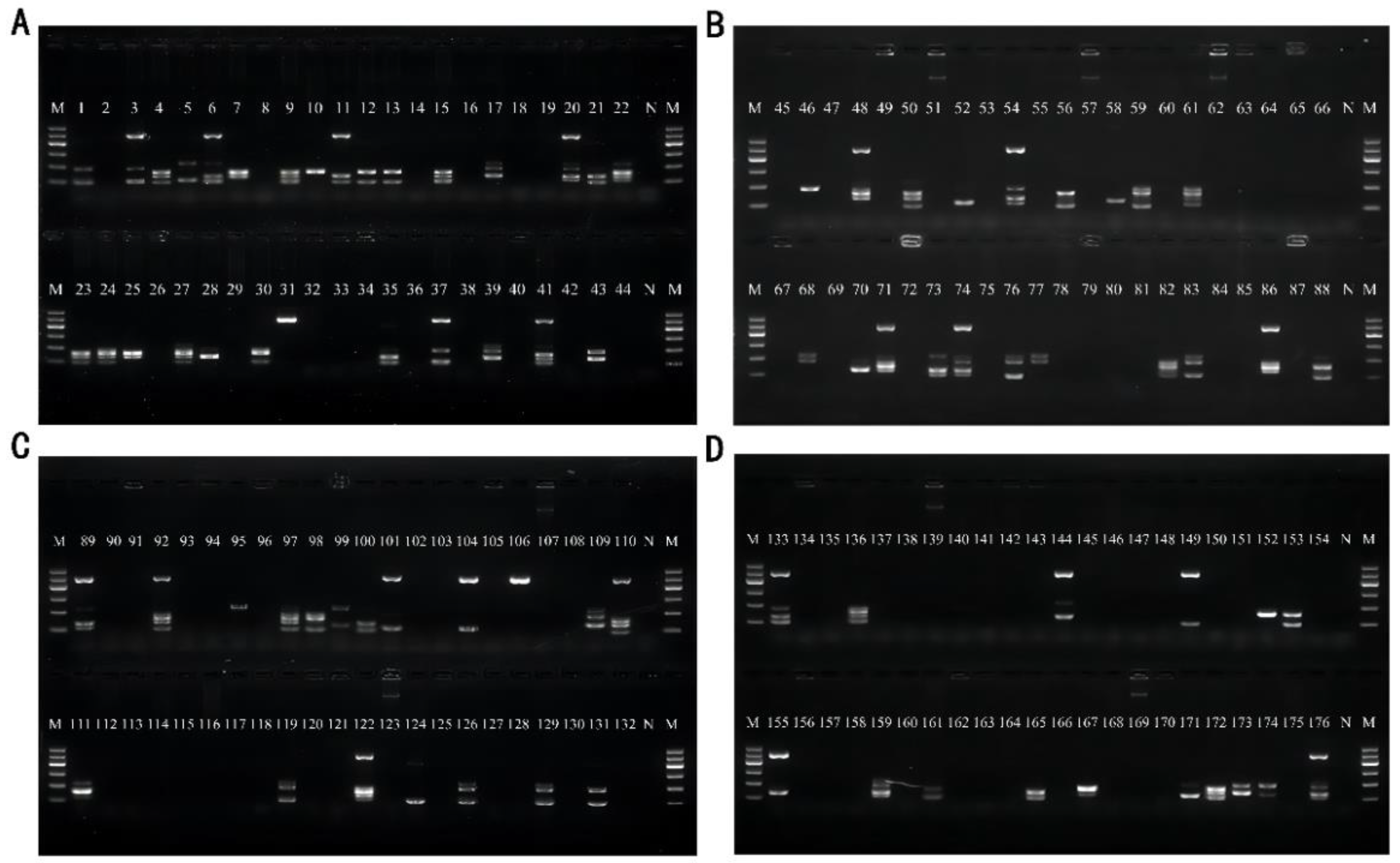
3.5. Artificial Contamination Sample Validation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Su, Y.-C.; Liu, C. Vibrio parahaemolyticus: A concern of seafood safety. Food Microbiol. 2007, 24, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Huss, H.H.; Jørgensen, L.V.; Vogel, B.F. Control options for Listeria monocytogenes in seafoods. Int. J. Food Microbiol. 2000, 62, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Pennington, H. Escherichia coli O157. Lancet 2010, 376, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Beuchat, L.R.; Kim, H.; Gurtler, J.B.; Lin, L.-C.; Ryu, J.-H.; Richards, G.M. Cronobacter sakazakii in foods and factors affecting its survival, growth, and inactivation. Int. J. Food Microbiol. 2009, 136, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Jennison, A.V.; Verma, N.K. Shigella flexneri infection: Pathogenesis and vaccine development. FEMS Microbiol. Rev. 2004, 28, 43–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altinok, I.; Kayis, S.; Capkin, E. Pseudomonas putida infection in rainbow trout. Aquaculture 2006, 261, 850–855. [Google Scholar] [CrossRef]
- Jones, M.K.; Oliver, J.D. Vibrio vulnificus: Disease and Pathogenesis. Infect. Immun. 2009, 77, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs Slifka, K.M.; Newton, A.E.; Mahon, B.E. Vibrio alginolyticus infections in the USA, 1988–2012. Epidemiol. Infect. 2017, 145, 1491–1499. [Google Scholar] [CrossRef] [Green Version]
- Gan, C.; Hu, J.; Cao, Q.; Zhao, R.; Li, Y.; Wang, Z.; Tao, Y.; Mo, X. Rapid identification of pathogens involved in pediatric osteoarticular infections by multiplex PCR. Ann. Transl. Med. 2020, 8, 203. [Google Scholar] [CrossRef]
- Sun, Q.; Cheng, J.; Lin, R.; Li, J.; Zhang, Y.; Liang, X.; Su, Y.; Pang, R.; Xue, L.; Zeng, H.; et al. A novel multiplex PCR method for simultaneous identification of hypervirulent Listeria monocytogenes clonal complex 87 and CC88 strains in China. Int. J. Food Microbiol. 2022, 366, 109558. [Google Scholar] [CrossRef]
- Hernandez Hernandez, O.; Gutierrez-Escolano, A.L.; Cancio-Lonches, C.; Iturriaga, M.H.; Pacheco-Aguilar, J.R.; Morales-Rayas, R.; Arvizu-Medrano, S.M. Multiplex PCR method for the detection of human norovirus, Salmonella spp., Shigella spp., and shiga toxin producing Escherichia coli in blackberry, coriander, lettuce and strawberry. Food Microbiol. 2022, 102, 103926. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, D.; Li, H.; Pang, J.; Guo, H.; Qiu, J. Establishment and Application of Multiplex PCR for Simultaneously Detecting Escherichia coli, Salmonella, Klebsiella pneumoniae, and Staphylococcus aureus in Minks. Front. Vet. Sci. 2020, 7, 588173. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.; Imran, M.; Yaqub, T.; Tayyab, M.; Shehzad, W.; Thomson, P.C. A novel multiplex PCR assay for simultaneous detection of nine clinically significant bacterial pathogens associated with bovine mastitis. Mol. Cell Probes. 2017, 33, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.G.; Wang, M.X.; Song, P.; Mao, S.; Wang, Y.; Yang, Y.; Luo, J.; Ren, S.; Zhang, D.Y. Designing highly multiplex PCR primer sets with Simulated Annealing Design using Dimer Likelihood Estimation (SADDLE). Nat. Commun. 2022, 13, 1881. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, C.; Okuda, J.; Ishibashi, M.; Iwanaga, M.; Garg, P.; Rammamurthy, T.; Wong, H.-C.; Depaola, A.; Kim, Y.B.; Albert, M.J.; et al. Pandemic Spread of an O3:K6 Clone of Vibrio parahaemolyticus and Emergence of Related Strains Evidenced by Arbitrarily Primed PCR and toxRS Sequence Analyses. J. Clin. Microbiol. 2000, 38, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Pauliello, R.J. Avaliação do Efeito Inibitório e da Expressão de Genes Associados à Resistência de Listeria Monocytogenes Exposta à Nisina nas Formas Livre e Nanoencapsulada em Lipossomos de Fosfatidilcolina. 2021. Available online: https://www.lume.ufrgs.br/handle/10183/233076 (accessed on 1 September 2022).
- Simons, A.; Alhanout, K.; Duval, R.E. Bacteriocins, Antimicrobial Peptides from Bacterial Origin: Overview of Their Biology and Their Impact against Multidrug-Resistant Bacteria. Microorganisms 2020, 8, 639. [Google Scholar] [CrossRef] [PubMed]
- Franz, E.; Klerks, M.M.; Vos, O.J.D.; Termorshuizen, A.J.; Bruggen, A.H.C. Prevalence of Shiga Toxin-Producing Escherichia coli stx1, stx2, eaeA, and rfbE Genes and Survival of E. coli O157:H7 in Manure from Organic and Low-Input Conventional Dairy Farms. Appl. Environ. Microbiol. 2007, 73, 2180–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin, N.Y.; Mulchandani, A.; Chen, W. Use of real-time polymerase chain reaction and molecular beacons for the detection of Escherichia coli O157:H7. Anal. Biochem. 2001, 289, 281–288. [Google Scholar] [CrossRef]
- Kuhnert, P.; Korczak, B.M.; Stephan, R.; Joosten, H.; Iversen, C. Phylogeny and prediction of genetic similarity of Cronobacter and related taxa by multilocus sequence analysis (MLSA). Int. J. Food Microbiol. 2009, 136, 152–158. [Google Scholar] [CrossRef]
- Vu, D.T.; Sethabutr, O.; Von Seidlein, L.; Tran, V.T.; Do, G.C.; Bui, T.C.; Le, H.T.; Lee, H.; Houng, S.; Hale, T.L.; et al. Detection of Shigella by a PCR assay targeting the ipaH gene suggests increased prevalence of shigellosis in Nha Trang, Vietnam. J. Clin. Microbiol. 2004, 42, 2031–2035. [Google Scholar]
- Ercolini, D.; Russo, F.; Blaiotta, G.; Pepe, O.; Mauriello, G.; Villani, F. Simultaneous Detection of Pseudomonas fragi, P. lundensis, and P. putida from Meat by Use of a Multiplex PCR Assay Targeting the carA Gene. Appl. Environ. Microbiol. 2007, 73, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Qiu, L.; Zhao, X.; Zhang, H.; Wang, L.; Hou, Z.; Gao, H.; Song, L. Variation analysis of pathogenic Vibrio spp. and Pseudomonas spp. in Changhai mollusc farming waters using real-time PCR assay during 2011–2014. Mar. Biol. Res. 2016, 12, 146–157. [Google Scholar] [CrossRef]
- Panicker, G.; Myers, M.L.; Bej, A.K. Rapid Detection of Vibrio vulnificus in Shellfish and Gulf of Mexico Water by Real-Time PCR. Appl. Environ. Microbiol. 2004, 70, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Hou, Z.; Li, N.; Qin, Q. Development of a SYBR Green I real-time PCR for quantitative detection of Vibrio alginolyticus in seawater and seafood. J. Appl. Microbiol. 2007, 103, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Jett, B.D.; Hatter, K.L.M.; Huycke, M.; Gilmore, M.S. Simplified Agar Plate Method for Quantifying Viable Bacteria. BioTechniques 1997, 23, 648–650. [Google Scholar] [CrossRef]
- Wen, Y.; Tan, Y.; Zhao, L.; Lv, X.; Lin, L.; Liang, D.; Wang, L. Rapid on-site detection of viable Escherichia coli O157: H7 in lettuce using immunomagnetic separation combined with PMAxx-LAMP and nucleic acid lateral flow strip. Microchem. J. 2022, 178, 107348. [Google Scholar] [CrossRef]
- Lv, X.; Wang, L.; Zhang, J.; He, X.; Shi, L.; Zhao, L. Quantitative detection of trace VBNC Cronobacter sakazakii by immunomagnetic separation in combination with PMAxx-ddPCR in dairy products. Food Microbiol. 2021, 99, 103831. [Google Scholar] [CrossRef]
- Henegariu, O.; Heerema, N.A.; Dlouhy, S.R.; Vance, G.H.; Vogt, P.H. Multiplex PCR: Critical parameters and step-by-step protocol. Biotechniques 1997, 23, 504–511. [Google Scholar] [CrossRef]
- Leander, B.S.; Lloyd, S.A.J.; Marshall, W.; Landers, S.C. Phylogeny of Marine Gregarines (Apicomplexa)—Pterospora, Lithocystis and Lankesteria—And the Origin(s) of Coelomic Parasitism. Protist 2006, 157, 45–60. [Google Scholar] [CrossRef]
- Nogva, H.K.; Rudi, K.; Naterstad, K.; Holck, A.; Lillehaug, D. Application of 5′-nuclease PCR for quantitative detection of Listeria monocytogenes in pure cultures, water, skim milk, and unpasteurized whole milk. Appl. Environ. Microbiol. 2000, 66, 4266–4271. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.N.; Bej, A.K. Detection of Vibrio parahaemolyticus in Shellfish by Use of Multiplexed Real-Time PCR with TaqMan Fluorescent Probes. Appl. Environ. Microbiol. 2006, 72, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gentry, T.J.; Grass, G.; Josephson, K.; Rensing, C.; Pepper, I.L. Real-time PCR quantification of a green fluorescent protein-labeled, genetically engineered Pseudomonas putida strain during 2-chlorobenzoate degradation in soil. FEMS Microbiol. Lett. 2004, 233, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.T.; Kim, Y.-R.; Kim, E.-Y.; Lee, J.-M.; Kong, I.-S. Detection of Vibrio cholerae and Vibrio vulnificus by duplex PCR specific to the groEL gene. Fish. Sci. 2013, 79, 335–340. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, M.; Wang, Q.; Cao, B.; He, X.; Li, K.; Feng, L.; Wang, L. Genetic Analysis of the Cronobacter sakazakii O4 to O7 O-Antigen Gene Clusters and Development of a PCR Assay for Identification of All C. sakazakii O Serotypes. Appl. Environ. Microbiol. 2012, 78, 3966–3974. [Google Scholar] [CrossRef] [Green Version]
- Ojha, S.C.; Yean Yean, C.; Ismail, A.; Banga Singh, K.-K. A Pentaplex PCR Assay for the Detection and Differentiation of Shigella Species. BioMed. Res. Int. 2013, 2013, 412370. [Google Scholar] [CrossRef] [Green Version]
- Jing-jing, Z.; Chang, C.; Peng, L.; Chun-hua, R.; Xiao, J.; Zhe, Z.; Chao-qun, H. SYBR Green I-based real-time PCR targeting the rpoX gene for sensitive and rapid detection of Vibrio alginolyticus. Mol. Cell. Probes 2011, 25, 137–141. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Bacterial Species | Source |
|---|---|---|
| Target Strains | ||
| 1 | Vibrio parahaemolyticus | ATCC 17802 |
| 2 | Listeria monocytogenes | ATCC 19115 |
| 3 | Cronobacter sakazakii | ATCC 35150 |
| 4 | Shigella flexneri | ATCC 12022 |
| 5 | Pseudomonas putida | SCAUFC-1 |
| 6 | Escherichia coli O157: H7 | ATCC 29544 |
| 7 | Vibrio vulnificus | ATCC 27562 |
| 8 | Vibrio alginolyticus | ATCC 33707 |
| Non-Target Strains | ||
| 9 | Escherichia coli EPEC | CICC 10412 |
| 10 | Escherichia coli EAEC | CICC 24186 |
| 11 | Escherichia coli ETEC | CICC 10667 |
| 12 | Pseudomonas aeruginosa | CMCC(B) 10104 |
| 13 | Bacillus cereus | CMCC(B) 63303 |
| 14 | Yersinia enterocolitica-1 | CICC 10869 |
| 15 | Yersinia enterocolitica-2 | CMCC(B) 52204 |
| 16 | Streptococcus pyogenes | IQCC 22107 |
| 17 | Salmonella enterica | ATCC 14028 |
| 18 | Vibrio cholerae (Vbo) | FSCC 232004 |
| 19 | Vibrio minima | ATCC 33653 |
| 20 | Staphylococcus aureus | CMCC(B)26003 |
| 21 | Klebsiella pneumoniae | SCAUFC-2 |
| Bacteria | Target Genes | Primer Sequences (5′-3′) | Product Size (bp) |
|---|---|---|---|
| V. parahaemolyticus | toxS | F-TTTTGGCCGTATCTATCCTT | 89 |
| R-CTGCCATTCATTTGATGTAAGC | |||
| L. monocytogenes | vir.R | F-GTCTAGTAGAAAAGAAGAAGTCC | 162 |
| R-GGTTTCCCAGGAAGTTTG | |||
| E. coli O157:H7 | rfbE | F-AAAACACTTTATGACCGTTGT | 110 |
| R-GCGCAGATATTTGTCATCCT | |||
| C. sakazakii | recN | F-CATATGGGTTTCGGTCATCGC | 186 |
| R-GCTGATTTTCGATGAAGTGGACG | |||
| S. flexneri | ipaH | F-GAAAGCCTACCAGCCGTA | 140 |
| R-TCTTCGAGGATGATAGTGC | |||
| P. putida | CarA | F-AGGAAATCCTTACAGACCCT | 500 |
| R-CAGGATGTTCAGCTTGACG | |||
| V. vulnificus | vvhA | F-TCCGATCGTTGTTTGACCGTA | 228 |
| R-TTTGACTTGTTGTAATGTGGGTT | |||
| V. alginolyticus | gyrB | F-CATTCCTGAACTCTGGTGT | 122 |
| R-CGTTTTGTTGGTGTTTAGGT |
| Optimization of Amplification Conditions | |||
|---|---|---|---|
| Single-Duplex PCR Amplification | |||
| Steps | Temperature | Time | Number of Cycles |
| Initial denaturation | 95 °C | 3 min | 1 |
| Denaturation | 94 °C | 25 s | 30 |
| Annealing | 48–55 °C | 25 s | |
| Initial extension | 72 °C | 10 s | |
| Final extension | 72 °C | 5 min | 1 |
| Multiplex PCR Amplification | |||
| Steps | Temperature | Time | Number of Cycles |
| Initial denaturation | 95 °C | 3 min | 1 |
| Denaturation | 94 °C | 25 s | 25 |
| Annealing | 48–58 °C | 25 s | |
| Initial extension | 72 °C | 10 s | |
| Final extension | 72 °C | 5 min | 1 |
| Group | Primer Addition (μL) | |||||||
|---|---|---|---|---|---|---|---|---|
| toxS | vir.R | rfbE | recN | ipaH | CarA | vvhA | gyrB | |
| 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 2 | 1 | 1.5 | 1 | 0.5 | 1 | 1 | 1 | 1 |
| 3 | 1 | 2 | 1 | 0.5 | 0.5 | 1 | 1 | 1 |
| 4 | 1 | 2.5 | 1 | 0.5 | 0.5 | 1 | 1 | 0.5 |
| 5 | 1 | 1 | 1.5 | 0.5 | 1 | 1 | 1 | 1 |
| 6 | 1.5 | 2 | 1 | 0.5 | 1 | 1 | 1 | 1 |
| 7 | 1 | 3 | 1 | 0.5 | 0.5 | 1 | 0.5 | 0.5 |
| Contaminated Samples | Situation of Artificial Pollution (Positive/Total) | Multiplex PCR Assay (Positive/Total) | Detection Rate (%) | Detection Sensitivity (CFU/mL) |
|---|---|---|---|---|
| Basa catfish | 16/22 | 16/22 | 100 | 104 |
| Beltfish | 12/22 | 12/22 | 100 | 104 |
| River shrimps | 9/22 | 9/22 | 100 | 104 |
| Sea shrimps | 11/22 | 11/22 | 100 | 104 |
| Scallops | 12/22 | 12/22 | 100 | 104 |
| Oysters | 7/22 | 7/22 | 100 | 104 |
| Seaweed | 6/22 | 6/22 | 100 | 104 |
| Skunk cabbage | 10/22 | 10/22 | 100 | 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Feng, X.; Chen, B.; Wang, X.; Liang, Z.; Wang, L. The Detection of Foodborne Pathogenic Bacteria in Seafood Using a Multiplex Polymerase Chain Reaction System. Foods 2022, 11, 3909. https://doi.org/10.3390/foods11233909
Li P, Feng X, Chen B, Wang X, Liang Z, Wang L. The Detection of Foodborne Pathogenic Bacteria in Seafood Using a Multiplex Polymerase Chain Reaction System. Foods. 2022; 11(23):3909. https://doi.org/10.3390/foods11233909
Chicago/Turabian StyleLi, Pengzhen, Xiaoxuan Feng, Baiyan Chen, Xiaoying Wang, Zuyue Liang, and Li Wang. 2022. "The Detection of Foodborne Pathogenic Bacteria in Seafood Using a Multiplex Polymerase Chain Reaction System" Foods 11, no. 23: 3909. https://doi.org/10.3390/foods11233909
APA StyleLi, P., Feng, X., Chen, B., Wang, X., Liang, Z., & Wang, L. (2022). The Detection of Foodborne Pathogenic Bacteria in Seafood Using a Multiplex Polymerase Chain Reaction System. Foods, 11(23), 3909. https://doi.org/10.3390/foods11233909