
NMR Spectroscopy Identifies Chemicals in Cigarette Smoke Condensate That Impair Skeletal Muscle Mitochondrial Function

 ,
,  , ,
, , 

Abstract
:1. Introduction
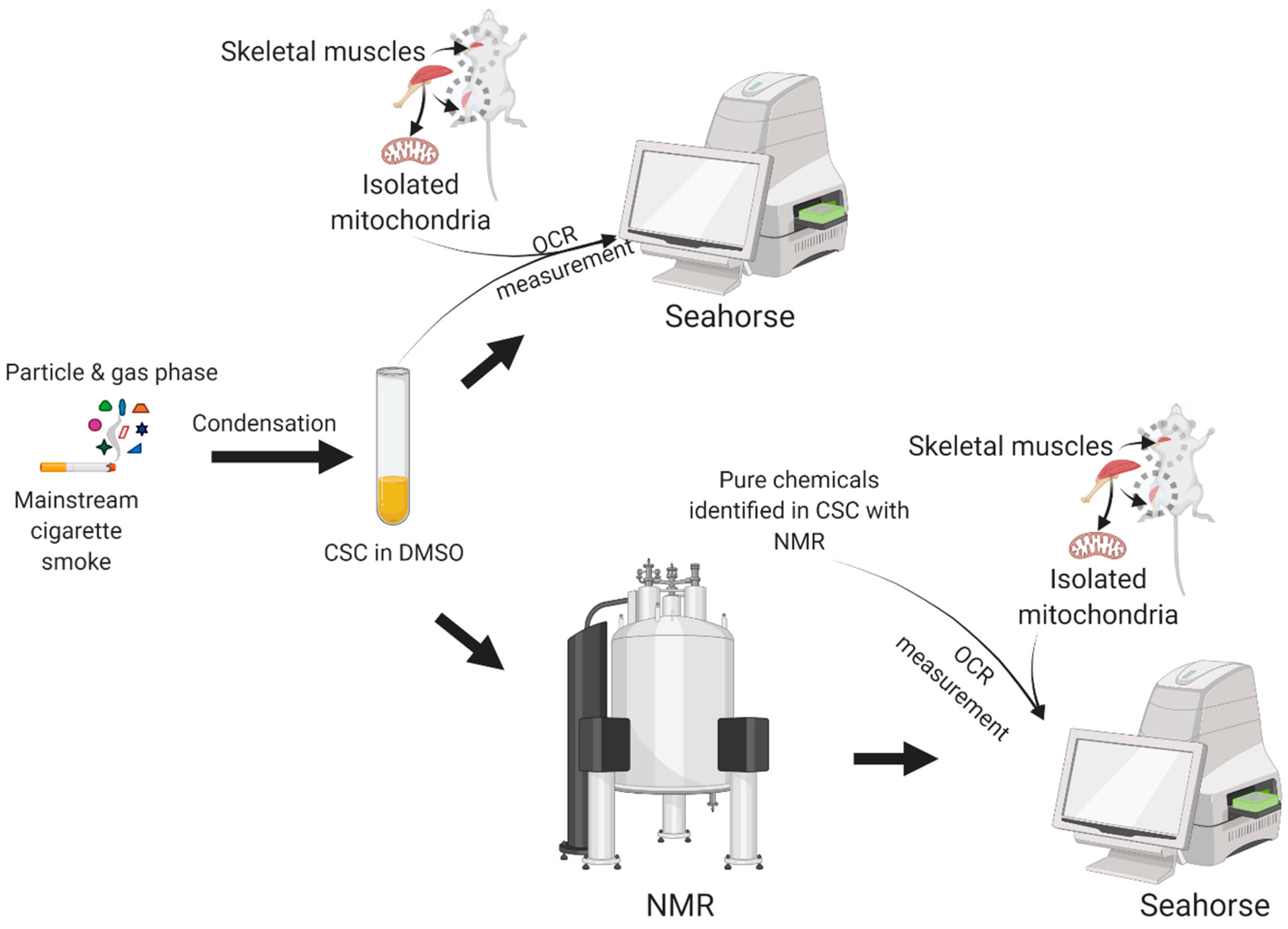
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Isolation of Skeletal Muscle Mitochondria
2.4. Seahorse XFe96 Assay: Oxygen Consumption by Skeletal Muscle Mitochondria
2.5. NMR Sample Preparation
2.6. Data Processing
2.7. Statistical Analysis
3. Results
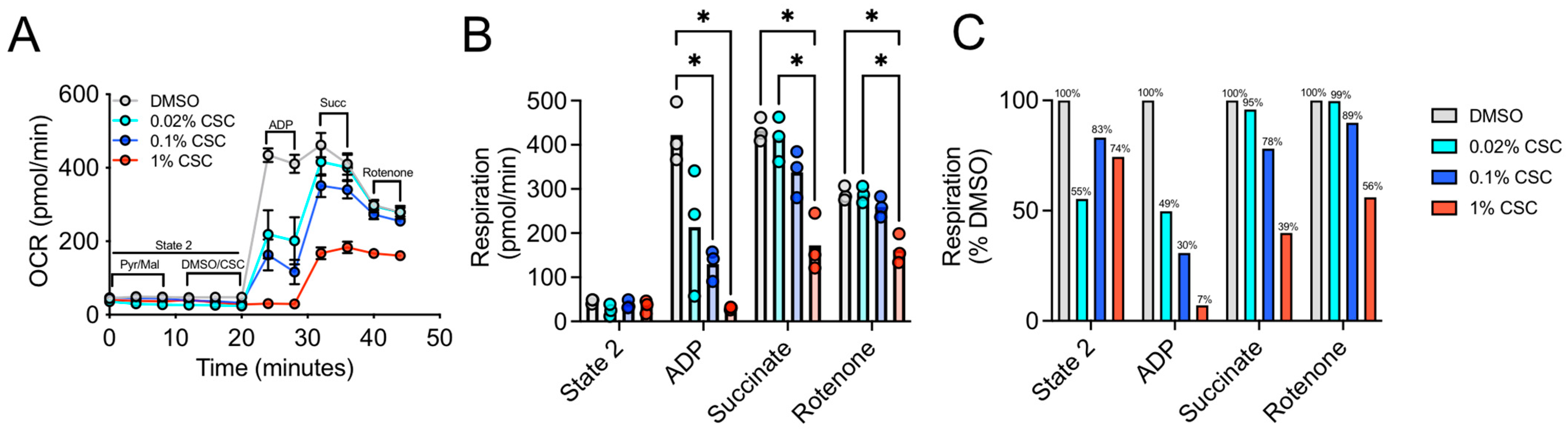
3.1. Acute Treatment with CSC Impairs Complex 1 Dependent Respiration in Skeletal Muscle Mitochondria
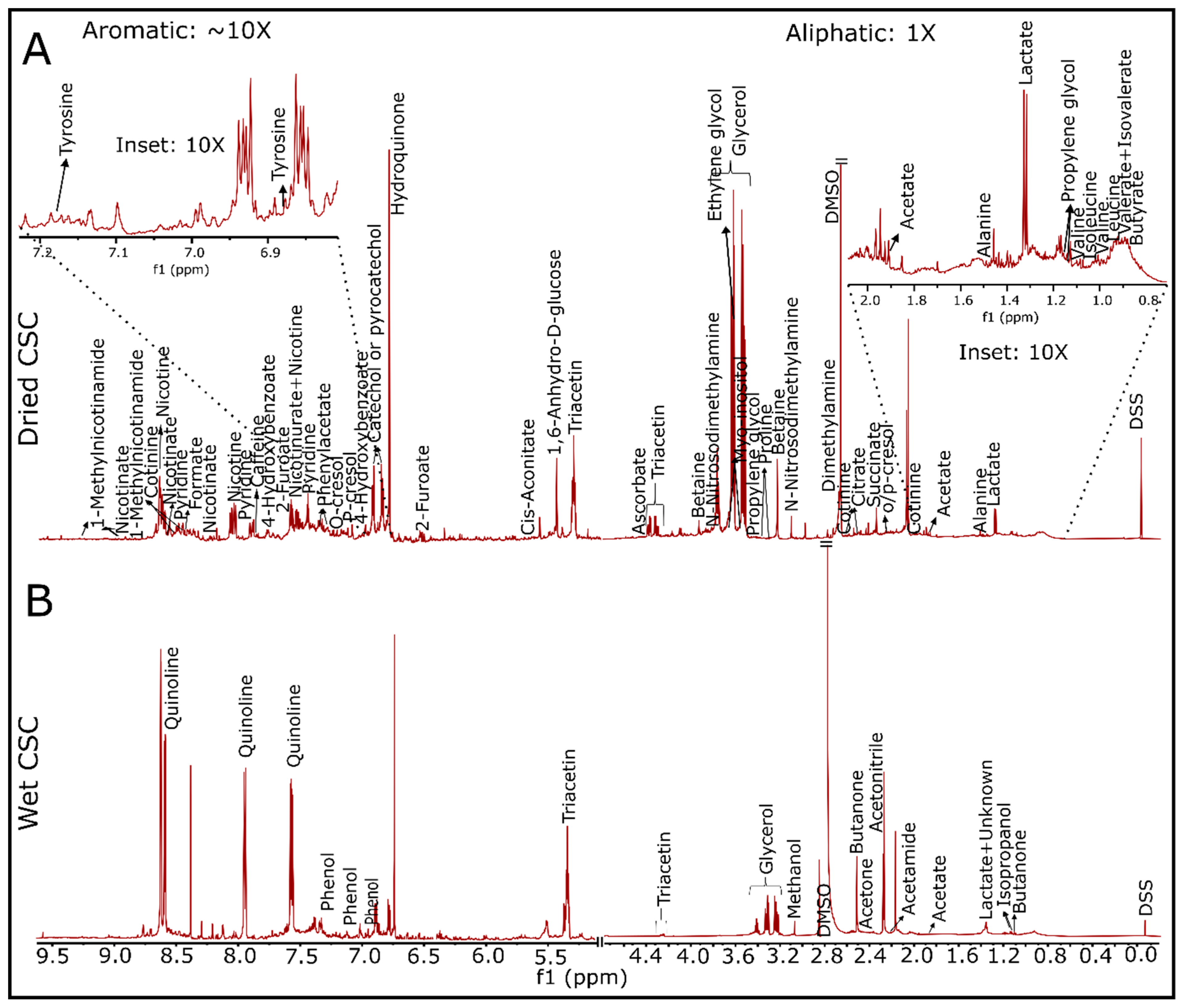
3.2. Detection of Water and Lipid Soluble Chemicals in 3R4F-Derived CSC via 1D/2D NMR
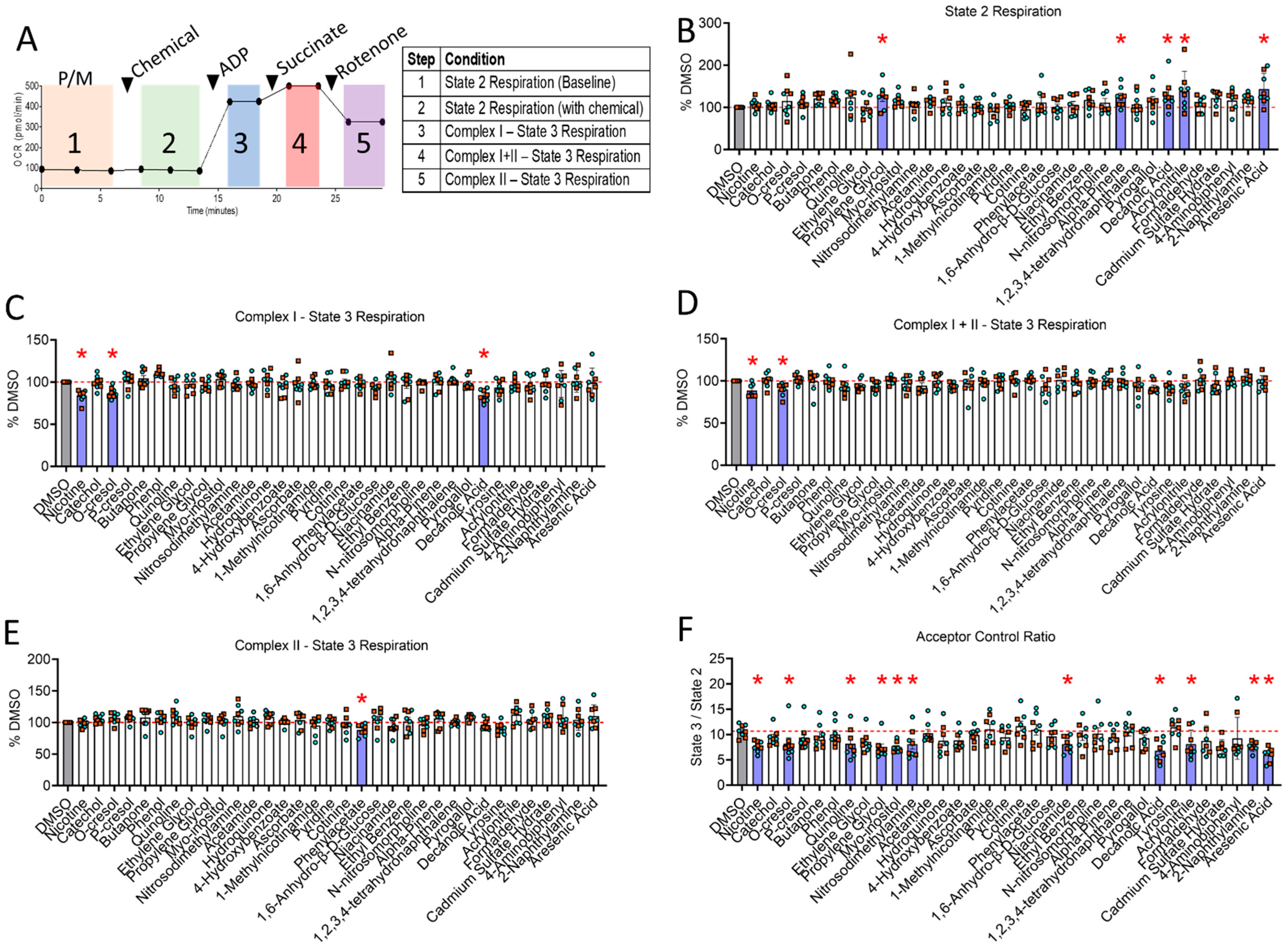
3.3. Mitochondrial Screening Identifies Nicotine, Decanoic Acid and o-Cresol as Toxins That Impair Complex I-Supported Mitochondrial Respiration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marquis, K.; Debigare, R.; Lacasse, Y.; LeBlanc, P.; Jobin, J.; Carrier, G.; Maltais, F. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Limpawattana, P.; Inthasuwan, P.; Putraveephong, S.; Boonsawat, W.; Theerakulpisut, D.; Sawanyawisuth, K. Sarcopenia in chronic obstructive pulmonary disease: A study of prevalence and associated factors in the Southeast Asian population. Chron. Respir. Dis. 2018, 15, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.E.; Maddocks, M.; Kon, S.S.; Canavan, J.L.; Nolan, C.M.; Clark, A.L.; Polkey, M.I.; Man, W.D. Sarcopenia in COPD: Prevalence, clinical correlates and response to pulmonary rehabilitation. Thorax 2015, 70, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitzman, D.W.; Nicklas, B.; Kraus, W.E.; Lyles, M.F.; Eggebeen, J.; Morgan, T.M.; Haykowsky, M. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1364–H1370. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.J.; Callahan, D.M.; Miller, M.S.; Tourville, T.W.; Hackett, S.B.; Couch, M.E.; Dittus, K. Skeletal muscle fiber size and fiber type distribution in human cancer: Effects of weight loss and relationship to physical function. Clin. Nutr. 2016, 35, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Maltais, F.; Decramer, M.; Casaburi, R.; Barreiro, E.; Burelle, Y.; Debigare, R.; Dekhuijzen, P.N.; Franssen, F.; Gayan-Ramirez, G.; Gea, J.; et al. An official American Thoracic Society/European Respiratory Society statement: Update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 189, e15–e62. [Google Scholar] [CrossRef] [Green Version]
- Orlander, J.; Kiessling, K.H.; Larsson, L. Skeletal muscle metabolism, morphology and function in sedentary smokers and nonsmokers. Acta Physiol. Scand. 1979, 107, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Orlander, J. Skeletal muscle morphology, metabolism and function in smokers and non-smokers. A study on smoking-discordant monozygous twins. Acta Physiol. Scand. 1984, 120, 343–352. [Google Scholar] [CrossRef]
- Degens, H.; Gayan-Ramirez, G.; van Hees, H.W. Smoking-induced skeletal muscle dysfunction: From evidence to mechanisms. Am. J. Respir. Crit. Care Med. 2015, 191, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Adami, A.; Cao, R.; Porszasz, J.; Casaburi, R.; Rossiter, H.B. Reproducibility of NIRS assessment of muscle oxidative capacity in smokers with and without COPD. Respir. Physiol. Neurobiol. 2017, 235, 18–26. [Google Scholar] [CrossRef]
- Boutagy, N.E.; Rogers, G.W.; Pyne, E.S.; Ali, M.M.; Hulver, M.W.; Frisard, M.I. Using Isolated Mitochondria from Minimal Quantities of Mouse Skeletal Muscle for High throughput Microplate Respiratory Measurements. J. Vis. Exp. 2015, 105, e53216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetterman, J.L.; Sammy, M.J.; Ballinger, S.W. Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology 2017, 391, 18–33. [Google Scholar] [CrossRef]
- Perez-Rial, S.; Barreiro, E.; Fernandez-Acenero, M.J.; Fernandez-Valle, M.E.; Gonzalez-Mangado, N.; Peces-Barba, G. Early detection of skeletal muscle bioenergetic deficit by magnetic resonance spectroscopy in cigarette smoke-exposed mice. PLoS ONE 2020, 15, e0234606. [Google Scholar] [CrossRef] [PubMed]
- Decker, S.T.; Kwon, O.S.; Zhao, J.; Hoidal, J.R.; Heuckstadt, T.; Richardson, R.S.; Sanders, K.A.; Layec, G. Skeletal muscle mitochondrial adaptations induced by long-term cigarette smoke exposure. Am. J. Physiol. Endoc. M 2021, 321, E80–E89. [Google Scholar] [CrossRef] [PubMed]
- Darabseh, M.Z.; Maden-Wilkinson, T.M.; Welbourne, G.; Wust, R.C.I.; Ahmed, N.; Aushah, H.; Selfe, J.; Morse, C.I.; Degens, H. Fourteen days of smoking cessation improves muscle fatigue resistance and reverses markers of systemic inflammation. Sci. Rep. UK 2021, 11, 12286. [Google Scholar] [CrossRef] [PubMed]
- Ajime, T.T.; Serre, J.; Wust, R.C.I.; Messa, G.A.M.; Poffe, C.; Swaminathan, A.; Maes, K.; Janssens, W.; Troosters, T.; Degens, H.; et al. Two Weeks of Smoking Cessation Reverse Cigarette Smoke-Induced Skeletal Muscle Atrophy and Mitochondrial Dysfunction in Mice. Nicotine Tob. Res. 2021, 23, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.K.; Fung, T.K.H.; Mak, J.C.W.; Cheung, S.Y.; He, W.J.; Leung, J.W.; Lau, B.W.M.; Ngai, S.P.C. The acute effects of cigarette smoke exposure on muscle fiber type dynamics in rats. PLoS ONE 2020, 15, e0233523. [Google Scholar] [CrossRef] [PubMed]
- Angenot, L. Chemical-composition of tobacco-smoke. J. Pharm. Belg. 1983, 38, 172–180. [Google Scholar]
- Barsanti, K.C.; Luo, W.; Isabelle, L.M.; Pankow, J.F.; Peyton, D.H. Tobacco smoke particulate matter chemistry by NMR. Magn. Reson. Chem. 2007, 45, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, J.; Neukomm, S. Current results of chemical studies of the composition of tobacco smoke. Oncologia 1957, 10, 124–129. [Google Scholar] [CrossRef]
- Borgerding, M.; Klus, H. Analysis of complex mixtures--cigarette smoke. Exp. Toxicol. Pathol. 2005, 57 (Suppl. S1), 43–73. [Google Scholar] [CrossRef]
- Borgerding, M.; Bodnar, J.; Curtin, G.; Swauger, J. The chemical composition of smokeless tobacco: A survey of products sold in the United States in 2006 and 2007. Regul. Toxicol. Pharmacol. 2012, 64, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, W.; Schlotzhauer, W.; Chortyk, O. Chemical-composition of nonsmoking tobacco products. J. Agric. Food Chem. 1988, 36, 48–50. [Google Scholar] [CrossRef]
- Dietrich, P.; Demole, E. Chemical composition of burley tobacco. Abstr. Pap. Am. Chem. Soc. 1977, 173, 7. [Google Scholar]
- Djulančić, N.; Radojičić, V.; Srbinovska, M. The influence of tobacco blend composition on carbon monoxide formation in mainstream cigarette smoke. Arh. Hig. Rada. Toksikol. 2013, 64, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eatough, D.; Benner, C.; Tang, H.; Landon, V.; Richards, G.; Caka, F.; Crawford, J.; Lewis, E.; Hansen, L.; Eatough, N. The chemical-composition of environmental tobacco-smoke 3. Identification of conservative tracers of environmental tobacco-smoke. Environ. Int. 1989, 15, 19–28. [Google Scholar] [CrossRef]
- Hecht, S.S.; Thorne, R.L.; Maronpot, R.R.; Hoffmann, D. A study of tobacco carcinogenesis. XIII. Tumor-promoting subfractions of the weakly acidic fraction. J. Natl. Cancer Inst. 1975, 55, 1329–1336. [Google Scholar] [CrossRef]
- Hecht, S.S.; Ornaf, R.M.; Hoffmann, D. Chemical studies on tobacco smoke. XXXIII. N′-nitrosonornicotine in tobacco: Analysis of possible contributing factors and biologic implications. J. Natl. Cancer Inst. 1975, 54, 1237–1244. [Google Scholar] [CrossRef]
- Hoffmann, D.; Wynder, E. Chemical composition and tumorigenicity of tobacco smoke. In The Chemistry of Tobacco and Tobacco Smoke; Schmeltz, I., Ed.; Plenum Press: New York, NY, USA, 1972; pp. 123–147. [Google Scholar]
- Margham, J.; McAdam, K.; Forster, M.; Liu, C.; Wright, C.; Mariner, D.; Proctor, C. Chemical Composition of Aerosol from an E-Cigarette: A Quantitative Comparison with Cigarette Smoke. Chem. Res. Toxicol. 2016, 29, 1662–1678. [Google Scholar] [CrossRef]
- Palic, R.; Stojanovic, G.; Alagic, S.; Nikolic, M.; Lepojevic, Z. Chemical composition and antimicrobial activity of the essential oil and CO2 extracts of the oriental tobacco, Prilep. Flavour Fragr. J. 2002, 17, 323–326. [Google Scholar] [CrossRef]
- Rodgman, A.; Smith, C.J.; Perfetti, T.A. The composition of cigarette smoke: A retrospective, with emphasis on polycyclic components. Hum. Exp. Toxicol. 2000, 19, 573–595. [Google Scholar] [CrossRef] [PubMed]
- Roemer, E.; Stabbert, R.; Rustemeier, K.; Veltel, D.J.; Meisgen, T.J.; Reininghaus, W.; Carchman, R.A.; Gaworski, C.L.; Podraza, K.F. Chemical composition, cytotoxicity and mutagenicity of smoke from US commercial and reference cigarettes smoked under two sets of machine smoking conditions. Toxicology 2004, 195, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.N.; Green, C.R.; Best, F.W.; Newell, M.P. Smoke composition. An extensive investigation of the water-soluble portion of cigarette smoke. J. Agric. Food Chem. 1977, 25, 310–320. [Google Scholar] [CrossRef]
- Stedman, R. Chemical Composition of Tobacco and Tobacco Smoke. Chem. Rev. 1968, 68, 153. [Google Scholar] [CrossRef]
- Ticha, J.; Wright, C. Rapid detection of toxic compounds in tobacco smoke condensates using high-resolution H-1-nuclear magnetic resonance spectroscopy. Anal. Methods 2016, 8, 6388–6397. [Google Scholar] [CrossRef] [Green Version]
- Maremanda, K.P.; Sundar, I.K.; Rahman, I. Role of inner mitochondrial protein OPA1 in mitochondrial dysfunction by tobacco smoking and in the pathogenesis of COPD. Redox Biol. 2021, 45, 102055. [Google Scholar] [CrossRef]
- Jia, G.Z.; Meng, Z.J.; Liu, C.H.; Ma, X.L.; Gao, J.; Liu, J.; Guo, R.; Yan, Z.Y.; Christopher, T.; Lopez, B.; et al. Nicotine induces cardiac toxicity through blocking mitophagic clearance in young adult rat. Life Sci. 2020, 257, 118084. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Itani, H.; Richmond, B.; Arslanbaeva, L.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension (vol 316, pg H639, 2019). Am. J. Physiol. Heart C 2019, 316, H939. [Google Scholar] [CrossRef] [PubMed]
- Leggett, A.; Wang, C.; Li, D.W.; Somogyi, A.; Bruschweiler-Li, L.; Bruschweiler, R. Identification of Unknown Metabolomics Mixture Compounds by Combining NMR, MS, and Cheminformatics. Methods Enzymol. 2019, 615, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Thome, T.; Salyers, Z.R.; Kumar, R.A.; Hahn, D.; Berru, F.N.; Ferreira, L.F.; Scali, S.T.; Ryan, T.E. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am. J. Physiol. Cell Physiol. 2019, 317, C701–C713. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Spangenburg, E.E.; Neufer, P.D.; McClung, J.M. Targeted Expression of Catalase to Mitochondria Protects Against Ischemic Myopathy in High-Fat Diet-Fed Mice. Diabetes 2016, 65, 2553–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Alleman, R.J.; Tsang, A.M.; Green, T.D.; Neufer, P.D.; Brown, D.A.; McClung, J.M. Mitochondrial therapy improves limb perfusion and myopathy following hindlimb ischemia. J. Mol. Cell Cardiol. 2016, 97, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanbakhsh, S.; Liu, P.; Bjorndahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Correction: Accurate, Fully-Automated NMR Spectral Profiling for Metabolomics. PLoS ONE 2015, 10, e0132873. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.E.; Khattri, R.B.; Guingab-Cagmat, J.; Camp, E.F.; Merritt, M.E.; Garrett, T.J.; Patterson, J.T. Metabolomic profiles differ among unique genotypes of a threatened Caribbean coral. Sci. Rep. 2019, 9, 6067. [Google Scholar] [CrossRef] [PubMed]
- Myer, C.; Abdelrahman, L.; Banerjee, S.; Khattri, R.B.; Merritt, M.E.; Junk, A.K.; Lee, R.K.; Bhattacharya, S.K. Aqueous humor metabolite profile of pseudoexfoliation glaucoma is distinctive. Mol. Omics. 2020, 16, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Myer, C.; Perez, J.; Abdelrahman, L.; Mendez, R.; Khattri, R.B.; Junk, A.K.; Bhattacharya, S.K. Differentiation of soluble aqueous humor metabolites in primary open angle glaucoma and controls. Exp. Eye Res. 2020, 194, 108024. [Google Scholar] [CrossRef] [PubMed]
- Osis, G.; Webster, K.; Harris, A.; Lee, H.; Chen, C.; Fang, L.; Romero, M.; Khattri, R.; Merritt, M.; Verlander, J.; et al. Regulation of renal NaDC1 expression and citrate excretion by NBCe1-A. Am. J. Physiol. Ren. Physiol. 2019, 317, F489–F501. [Google Scholar] [CrossRef] [PubMed]
- Mahrous, E.A.; Lee, R.B.; Lee, R.E. Lipid profiling using two-dimensional heteronuclear single quantum coherence NMR. Methods Mol. Biol. 2009, 579, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Marion, D.; Wüthrich, K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of (1)H-(1)H spin-spin coupling constants in proteins. 1983. Biochem. Biophys Res. Commun. 2012, 425, 519–526. [Google Scholar] [CrossRef]
- Clendinen, C.S.; Lee-McMullen, B.; Williams, C.M.; Stupp, G.S.; Vandenborne, K.; Hahn, D.A.; Walter, G.A.; Edison, A.S. ¹³C NMR metabolomics: Applications at natural abundance. Anal. Chem. 2014, 86, 9242–9250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaraja, M.; Turner, C.; Souza, K.; Singer, S. Ex vivo two-dimensional proton nuclear magnetic resonance spectroscopy of smooth muscle tumors: Advantages of total correlated spectroscopy over homonuclear J-correlated spectroscopy. Cancer Res. 1994, 54, 6037–6040. [Google Scholar] [PubMed]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Chen, C.C.; Lee, H. Genotoxicity and DNA adduct formation of incense smoke condensates: Comparison with environmental tobacco smoke condensates. Mutat. Res. 1996, 367, 105–114. [Google Scholar] [CrossRef]
- Forbes, W.F.; Robinson, J.C.; Wright, G.F. Free radicals of biological interest. I. Electron spin resonance spectra of tobacco smoke condensates. Can. J. Biochem. 1967, 45, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Hannan, M.A.; Estes, R.S.; Hurley, L.H. Induction and potentiation of lethal and genetic effects of ultraviolet light by tobacco smoke condensates in yeast. Environ. Res. 1980, 21, 97–107. [Google Scholar] [CrossRef]
- Maertens, R.M.; White, P.A.; Rickert, W.; Levasseur, G.; Douglas, G.R.; Bellier, P.V.; McNamee, J.P.; Thuppal, V.; Walker, M.; Desjardins, S. The genotoxicity of mainstream and sidestream marijuana and tobacco smoke condensates. Chem. Res. Toxicol. 2009, 22, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Misfeld, J.; Weber, K.H. Animal experiments with tobacco smoke condensates and their statistical evaluation. Planta Med. 1972, 22, 281–292. [Google Scholar] [CrossRef]
- Nguyen Van Binh, P.; Zhou, D.; Baudouin, F.; Martin, C.; Radionoff, M.; Dutertre, H.; Marchand, V.; Thevenin, M.; Warnet, J.M.; Thien Duc, H. Modulation of the primary and the secondary antibody response by tobacco smoke condensates. Biomed. Pharmacother. 2004, 58, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.R.; Estefan, R.M.; Gause, E.M.; Montalvo, D.A. An electron spin resonance study of tobacco smoke condensates and their effects upon blood constituents. Environ. Res. 1968, 2, 47–71. [Google Scholar] [CrossRef]
- Schmähl, D. Comparative studies in rats of the carcinogenic effect of different tobacco extracts and tobacco smoke condensates. Arzneimittelforschung 1968, 18, 814–817. [Google Scholar] [PubMed]
- Schmähl, D. Quantitative investigations of carcinogenic effects of tobacco smoke condensates in rats. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 1971, 76, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Wehner, F.C.; van Rensburg, S.J.; Thiel, P.G. Mutagenicity of marijuana and Transkei tobacco smoke condensates in the Salmonella/microsome assay. Mutat Res. 1980, 77, 135–142. [Google Scholar] [CrossRef]
- Spruit, M.A.; Singh, S.J.; Garvey, C.; ZuWallack, R.; Nici, L.; Rochester, C.; Hill, K.; Holland, A.E.; Lareau, S.C.; Man, W.D.; et al. An official American Thoracic Society/European Respiratory Society statement: Key concepts and advances in pulmonary rehabilitation. Am. J. Respir. Crit. Care Med. 2013, 188, e13–e64. [Google Scholar] [CrossRef]
- Jaccard, G.; Tafin Djoko, D.; Moennikes, O.; Jeannet, C.; Kondylis, A.; Belushkin, M. Comparative assessment of HPHC yields in the Tobacco Heating System THS2.2 and commercial cigarettes. Regul. Toxicol. Pharmacol. 2017, 90, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.M.; Chambers, D.M.; Pazo, D.Y.; Moliere, F.; Blount, B.C.; Watson, C.H. Simultaneous analysis of 22 volatile organic compounds in cigarette smoke using gas sampling bags for high-throughput solid-phase microextraction. Anal. Chem. 2014, 86, 7088–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adami, A.; Corvino, R.B.; Calmelat, R.A.; Porszasz, J.; Casaburi, R.; Rossiter, H.B. Muscle Oxidative Capacity Is Reduced in Both Upper and Lower Limbs in COPD. Med. Sci. Sport Exer. 2020, 52, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, R.M.; Hoff, J.; Wagner, P.D.; Richardson, R.S. Determinants of the diminished exercise capacity in patients with chronic obstructive pulmonary disease: Looking beyond the lungs. J. Physiol. Lond. 2020, 598, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.R.; Trinity, J.D.; Kwon, O.S.; Layec, G.; Garten, R.S.; Park, S.Y.; Nelson, A.D.; Richardson, R.S. Altered skeletal muscle mitochondrial phenotype in COPD: Disease vs. disuse. J. Appl. Physiol. 2018, 124, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Q.; Long, X.Y.; Xie, Y.; Zhao, Z.H.; Fang, L.Z.; Liu, L.; Fu, W.P.; Shu, J.K.; Wu, J.H.; Dai, L.M. Relationship between PPARa mRNA expression and mitochondrial respiratory function and ultrastructure of the skeletal muscle of patients with COPD. Bioengineered 2017, 8, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.R.; Trinity, J.D.; Layec, G.; Garten, R.S.; Park, S.Y.; Rossman, M.J.; Larsen, S.; Dela, F.; Richardson, R.S. Quadriceps exercise intolerance in patients with chronic obstructive pulmonary disease: The potential role of altered skeletal muscle mitochondrial respiration. J. Appl. Physiol. 2015, 119, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente-Maestu, L.; Perez-Parra, J.; Godoy, R.; Moreno, N.; Tejedor, A.; Gonzalez-Aragoneses, F.; Bravo, J.L.; Alvarez, F.V.; Camano, S.; Agusti, A. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur. Respir. J. 2009, 33, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; Godin, R.; Sinnreich, M.; Baril, J.; Bourbeau, J.; Perrault, H.; Taivassalo, T.; Burelle, Y. The Mitochondrial Phenotype of Peripheral Muscle in Chronic Obstructive Pulmonary Disease Disuse or Dysfunction? Am. J. Resp. Crit. Care 2008, 178, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoshnazar, M.; Bigdeli, M.R.; Parvardeh, S.; Pouriran, R. Attenuating effect of alpha-pinene on neurobehavioural deficit, oxidative damage and inflammatory response following focal ischaemic stroke in rat. J. Pharm. Pharmacol. 2019, 71, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormier, A.; Morin, C.; Zini, R.; Tillement, J.P.; Lagrue, G. In vitro effects of nicotine on mitochondrial respiration and superoxide anion generation. Brain Res. 2001, 900, 72–79. [Google Scholar] [CrossRef]
- Malińska, D.; Więckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymański, J.; Mathis, C.; Van der Toorn, M.; et al. Mitochondria as a possible target for nicotine action. J. Bioenerg. Biomembr. 2019, 51, 259–276. [Google Scholar] [CrossRef] [Green Version]
- Cormier, A.; Morin, C.; Zini, R.; Tillement, J.P.; Lagrue, G. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology 2003, 44, 642–652. [Google Scholar] [CrossRef]
- Das, S.; Gautam, N.; Dey, S.K.; Maiti, T.; Roy, S. Oxidative stress in the brain of nicotine-induced toxicity: Protective role of Andrographis paniculata Nees and vitamin E. Appl. Physiol. Nutr. Metab. 2009, 34, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Dewar, B.J.; Bradford, B.U.; Thurman, R.G. Nicotine increases hepatic oxygen uptake in the isolated perfused rat liver by inhibiting glycolysis. J. Pharmacol. Exp. Ther. 2002, 301, 930–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, A. Effects of cresols (o-, m-, and p-isomers) on the bioenergetic system in isolated rat liver mitochondria. Drug Chem. Toxicol. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- Doughmi, D.; Bennis, L.; Berrada, A.; Derkaoui, A.; Shimi, A.; Khatouf, M. Severe ARDS Complicating an Acute Intentional Cresol Poisoning. Case Rep. Crit. Care 2019, 2019, 6756352. [Google Scholar] [CrossRef] [PubMed]
- Fedotcheva, N.I.; Kazakov, R.E.; Kondrashova, M.N.; Beloborodova, N.V. Toxic effects of microbial phenolic acids on the functions of mitochondria. Toxicol. Lett. 2008, 180, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Moss, C.W.; Hatheway, C.L.; Lambert, M.A.; McCroskey, L.M. Production of phenylacetic and hydroxyphenylacetic acids by clostridium botulinum type G. J. Clin. Microbiol. 1980, 11, 743–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Westhoff, T.H.; Krauser, P.; Zidek, W.; van der Giet, M. The uraemic toxin phenylacetic acid increases the formation of reactive oxygen species in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2008, 23, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Loskovich, M.V.; Grivennikova, V.G.; Cecchini, G.; Vinogradov, A.D. Inhibitory effect of palmitate on the mitochondrial NADH:ubiquinone oxidoreductase (complex I) as related to the active-de-active enzyme transition. Biochem. J. 2005, 387, 677–683. [Google Scholar] [CrossRef]
- Luzikov, V.N.; Saks, V.A.; Berezin, I.V. Comparative study of thermal degradation of electron transfer particle and reconstituted respiratory chain. Relation of electron transfer to reactivation of submitochondrial particles. Biochim. Biophys. Acta 1970, 223, 16–30. [Google Scholar] [CrossRef]
- Hillered, L.; Chan, P.H. Effects of arachidonic acid on respiratory activities in isolated brain mitochondria. J. Neurosci. Res. 1988, 19, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Morii, H.; Tamura, M.; Hayaishi, O.; Watanabe, Y. A possible mechanism of mitochondrial dysfunction during cerebral ischemia: Inhibition of mitochondrial respiration activity by arachidonic acid. Arch Biochem. Biophys. 1991, 289, 33–38. [Google Scholar] [CrossRef]
- Batayneh, N.; Kopacz, S.J.; Lee, C.P. The modes of action of long chain alkyl compounds on the respiratory chain-linked energy transducing system in submitochondrial particles. Arch Biochem. Biophys. 1986, 250, 476–487. [Google Scholar] [CrossRef]
- Schewe, T.; Albracht, S.P.; Ludwig, P.; Rapoport, S.M. Two modes of irreversible inactivation of the mitochondrial electron-transfer system by tetradecanoic acid. Biochim. Biophys. Acta 1985, 807, 210–215. [Google Scholar] [CrossRef]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M.; Crimi, M.; Ghelli, A. Natural variation in the potency and binding sites of mitochondrial quinone-like inhibitors. Biochem. Soc. Trans. 1994, 22, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okun, J.G.; Lümmen, P.; Brandt, U. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase). J. Biol. Chem. 1999, 274, 2625–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Andersen, R.; Kasperbauer, M. Chemical composition of tobacco-leaves altered by near-ultraviolet and intensity of visible light. Plant Physiol. 1973, 51, 723–726. [Google Scholar] [CrossRef] [Green Version]
- Benner, C.; Bayona, J.; Caka, F.; Tang, H.; Lewis, L.; Crawford, J.; Lamb, J.; Lee, M.; Lewis, E.; Hansen, L.; et al. chemical-composition of environmental tobacco-smoke 2. particulate-phase compounds. Environ. Sci. Technol. 1989, 23, 688–699. [Google Scholar] [CrossRef]
- Adam, T.; Baker, R.R.; Zimmermann, R. Investigation, by single photon ionisation (SPI)-time-of-flight mass spectrometry (TOFMS), of the effect of different cigarette-lighting devices on the chemical composition of the first cigarette puff. Anal. Bioanal. Chem. 2007, 387, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Alagic, S.; Stancic, I.; Palic, R.; Stojanovic, G.; Nikolic, M. Chemical composition and antimicrobial activity of the essential oil of the oriental tobacco Yaka. J. Essent. Oil Res. 2002, 14, 230–232. [Google Scholar] [CrossRef]
- Alagic, S.; Stancic, I.; Palic, R.; Stojanovic, G.; Lepojevic, Z. Chemical composition of the supercritical CO2 extracts of the Yaka, Prilep and Otlja tobaccos. J. Essent. Oil Res. 2006, 18, 185–188. [Google Scholar] [CrossRef]
- Breheny, D.; Cunningham, F.; Kilford, J.; Payne, R.; Dillon, D.; Meredith, C. Application of a modified gaseous exposure system to the in vitro toxicological assessment of tobacco smoke toxicants. Environ. Mol. Mutagen. 2014, 55, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Eatough, D.; Benner, C.; Bayona, J.; Richards, G.; Lamb, J.; Lee, M.; Lewis, E.; Hansen, L. Chemical-composition of environmental tobacco-smoke.1. Gas-phase acids and bases. Environ. Sci. Technol. 1989, 23, 679–687. [Google Scholar] [CrossRef]
- Kamissoko, A.; Carré, V.; Schramm, S.; Aubriet, F. Study of the mainstream cigarette smoke aerosols by Fourier transform ion cyclotron resonance mass spectrometry coupled to laser/desorption and electrospray ionization—Additional insights on the heteroaromatic components. Rapid. Commun. Mass Spectrom. 2019, 33 (Suppl. S1), 95–108. [Google Scholar] [CrossRef]
- Demkowska, I.; Polkowska, Z.; Namieśnik, J. Application of ion chromatography for the determination of inorganic ions, especially thiocyanates in human saliva samples as biomarkers of environmental tobacco smoke exposure. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 875, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Famele, M.; Ferranti, C.; Abenavoli, C.; Palleschi, L.; Mancinelli, R.; Draisci, R. The chemical components of electronic cigarette cartridges and refill fluids: Review of analytical methods. Nicotine Tob. Res. 2015, 17, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Flicker, T.M.; Green, S.A. Comparison of gas-phase free-radical populations in tobacco smoke and model systems by HPLC. Environ. Health Perspect 2001, 109, 765–771. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, M.; Kong, H.; Cai, J.; Wu, J.; Wu, M.; Hua, R.; Liu, J.; Xu, G. Characterization of complex hydrocarbons in cigarette smoke condensate by gas chromatography-mass spectrometry and comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry. J. Chromatogr. A 2004, 1043, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Wachsmuth, C.; Buchholz, C.; Bentley, M. A complex matrix characterization approach, applied to cigarette smoke, that integrates multiple analytical methods and compound identification strategies for non-targeted liquid chromatography with high-resolution mass spectrometry. Rapid. Commun. Mass Spectrom. 2020, 34, e8571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, R.P.; Strongin, R.M.; Peyton, D.H. Solvent Chemistry in the Electronic Cigarette Reaction Vessel. Sci. Rep. 2017, 7, 42549. [Google Scholar] [CrossRef] [Green Version]
- Remaud, G.; Debon, A.; Martin, Y.; Martin, G.; Martin, G. Authentication of bitter almond oil and cinnamon oil: Application of the SNIF-NMR method to benzaldehyde. J. Agric. Food Chem. 1997, 45, 4042–4048. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Chemicals | Concentration (mM) | S.No. | Chemicals | Concentration (mM) |
|---|---|---|---|---|---|
| 1 | Quinoline | 9.22 | 18 | Propylene glycol | 0.14 |
| 2 | Decanoic acid | 9.0 | 19 | o-Cresol | 0.087 |
| 3 | Ethylene glycol | 2.12 | 20 | Ethyl benzene | 0.08 |
| 4 | Butanone | 1.46 | 21 | N-nitrosomorpholine | 0.07 |
| 5 | Pyrogallol | 1.07 | 22 | p-cresol | 0.06 |
| 6 | Phenol | 0.73 | 23 | Formaldehyde | 0.06 |
| 7 | Acetamide | 0.69 | 24 | Tyrosine | 0.04 |
| 8 | Myo-Inositol | 0.68 | 25 | Pyridine | 0.04 |
| 9 | Nicotine | 0.65 | 26 | Phenylacetate | 0.04 |
| 10 | 1,2,3,4-tetrahydronaphthalene | 0.64 | 27 | 4-Hydroxybenzoate | 0.03 |
| 11 | N-Nitrosodimethylamine | 0.59 | 28 | Niacinamide | 0.02 |
| 12 | 1,6-Anhydro-β-D-glucose | 0.47 | 29 | 1-Methylnicotinamide | 0.007 |
| 13 | Hydroquinone | 0.43 | 30 | Acrylonitrile | 0.76 |
| 14 | Catechol | 0.37 | 31 | 2-Naphthylamine | 0.0042 |
| 15 | α-pinene | 0.34 | 32 | Arsenic (III) chloride | 0.0016 |
| 16 | Ascorbate | 0.28 | 33 | Cadmium sulfate hydrate | 0.0016 |
| 17 | Cotinine | 0.183 | 34 | 4-Aminobiphenyl | 0.000061 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khattri, R.B.; Thome, T.; Fitzgerald, L.F.; Wohlgemuth, S.E.; Hepple, R.T.; Ryan, T.E. NMR Spectroscopy Identifies Chemicals in Cigarette Smoke Condensate That Impair Skeletal Muscle Mitochondrial Function. Toxics 2022, 10, 140. https://doi.org/10.3390/toxics10030140
Khattri RB, Thome T, Fitzgerald LF, Wohlgemuth SE, Hepple RT, Ryan TE. NMR Spectroscopy Identifies Chemicals in Cigarette Smoke Condensate That Impair Skeletal Muscle Mitochondrial Function. Toxics. 2022; 10(3):140. https://doi.org/10.3390/toxics10030140
Chicago/Turabian StyleKhattri, Ram B., Trace Thome, Liam F. Fitzgerald, Stephanie E. Wohlgemuth, Russell T. Hepple, and Terence E. Ryan. 2022. "NMR Spectroscopy Identifies Chemicals in Cigarette Smoke Condensate That Impair Skeletal Muscle Mitochondrial Function" Toxics 10, no. 3: 140. https://doi.org/10.3390/toxics10030140
APA StyleKhattri, R. B., Thome, T., Fitzgerald, L. F., Wohlgemuth, S. E., Hepple, R. T., & Ryan, T. E. (2022). NMR Spectroscopy Identifies Chemicals in Cigarette Smoke Condensate That Impair Skeletal Muscle Mitochondrial Function. Toxics, 10(3), 140. https://doi.org/10.3390/toxics10030140