Author Contributions
Conceptualization, A.R.F.; Data curation, A.R.F.; Formal analysis, A.R.F., J.W.P.-M., L.A.B.L., M.A.N., E.L.R., M.E.B., L.d.P.P., V.G.-J., P.A.F.W., P.C.M. and H.d.S.S.; Funding acquisition, A.R.F. and J.C.M.O.B.D.; Investigation, A.R.F., J.W.P.-M., G.M.F., E.L.R., M.E.B., L.d.P.P. and P.C.M.; Methodology, A.R.F., J.W.P.-M., L.A.B.L., M.A.N., E.L.R., M.E.B., L.d.P.P., P.A.F.W., P.C.M. and H.d.S.S.; Project administration, A.R.F., G.M.F., J.C.M.O.B.D. and H.d.S.S.; Resources, A.R.F., J.W.P.-M., L.A.B.L. and M.A.N.; Software, A.R.F. and P.A.F.W.; Supervision, A.R.F.; Validation, A.R.F. and L.A.B.L.; Visualization, A.R.F.; Writing—original draft, A.R.F., G.M.F. and J.C.M.O.B.D. All authors have read and agreed to the published version of the manuscript.
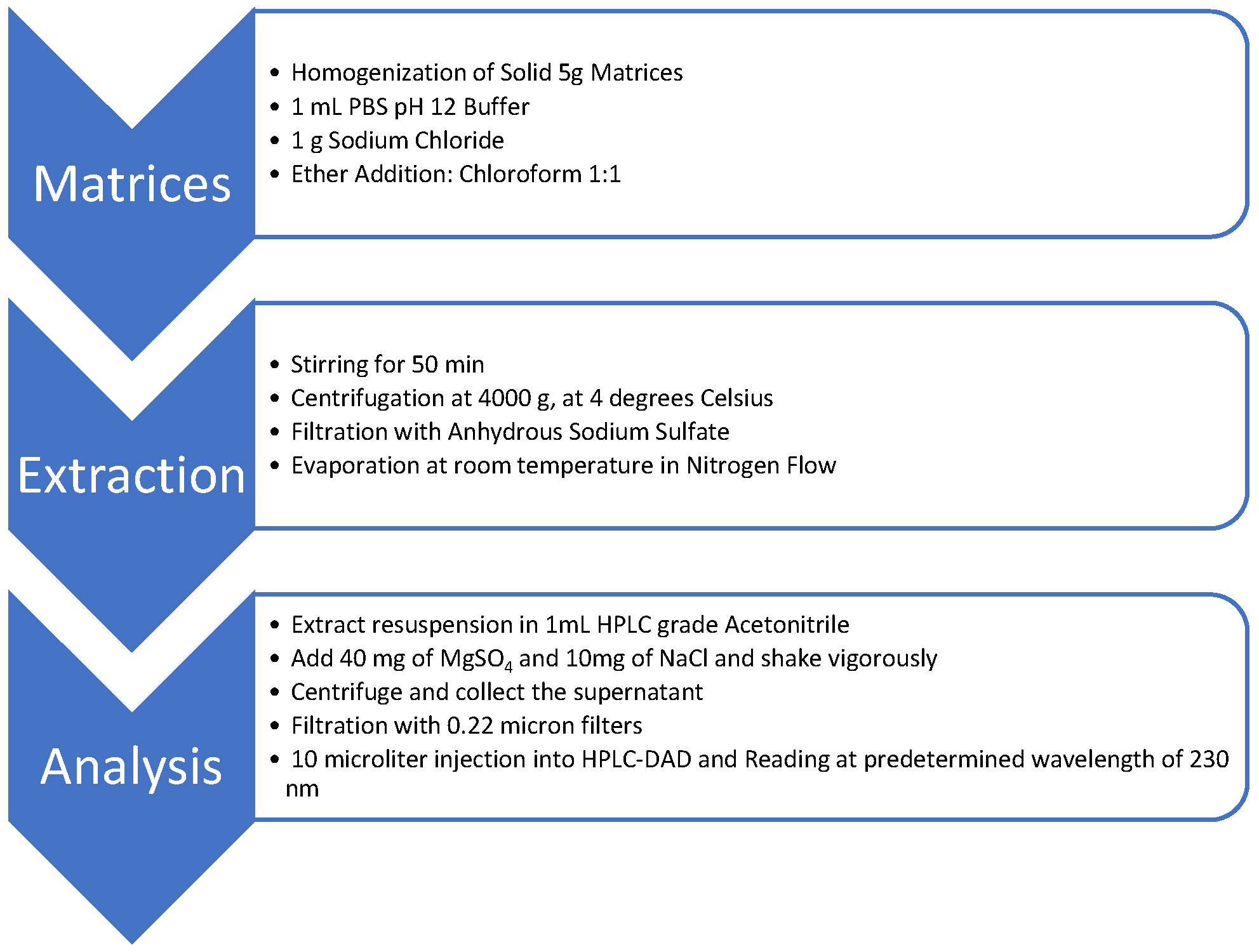
Figure 1.
Flowchart for extraction and preparation of samples for analysis.
Figure 1.
Flowchart for extraction and preparation of samples for analysis.
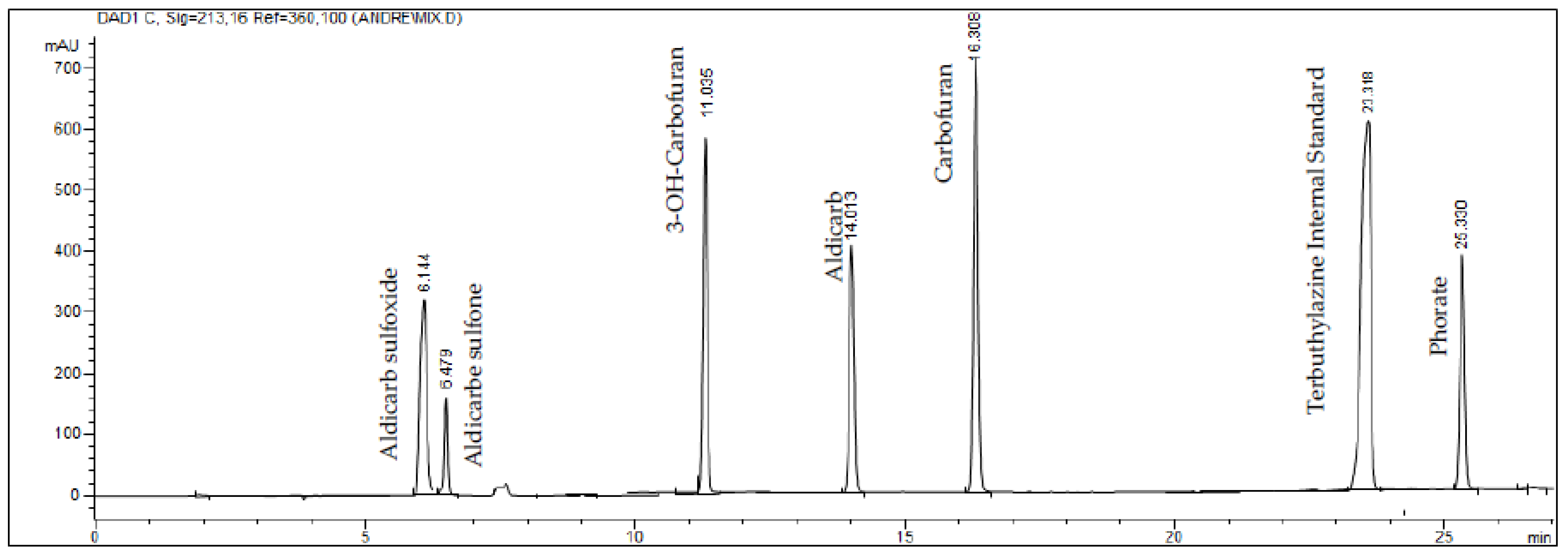
Figure 2.
Chromatogram of carbamates by HPLC–DAD. Simultaneous analysis of carbamates shows distinct separation of peaks that correspond to each tested carbamate. Notice the high resolution for each principal component and the absence of background noise.
Figure 2.
Chromatogram of carbamates by HPLC–DAD. Simultaneous analysis of carbamates shows distinct separation of peaks that correspond to each tested carbamate. Notice the high resolution for each principal component and the absence of background noise.
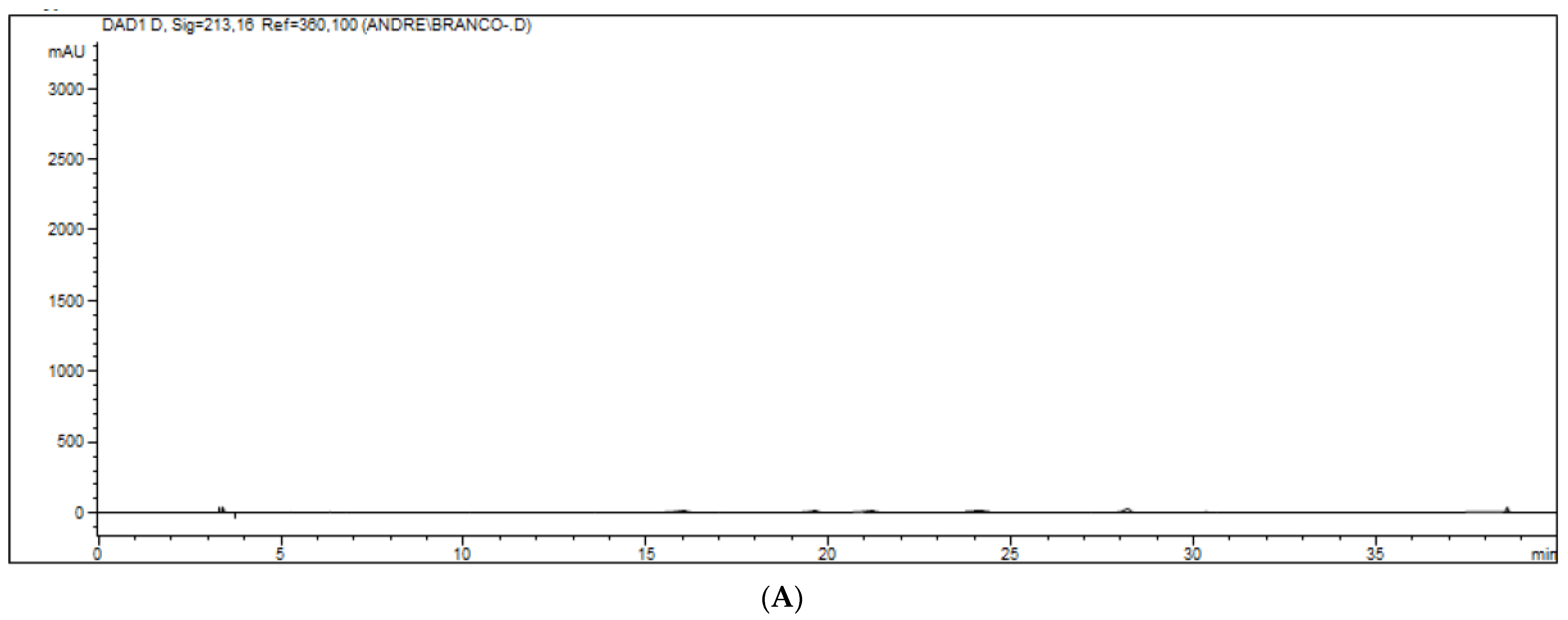
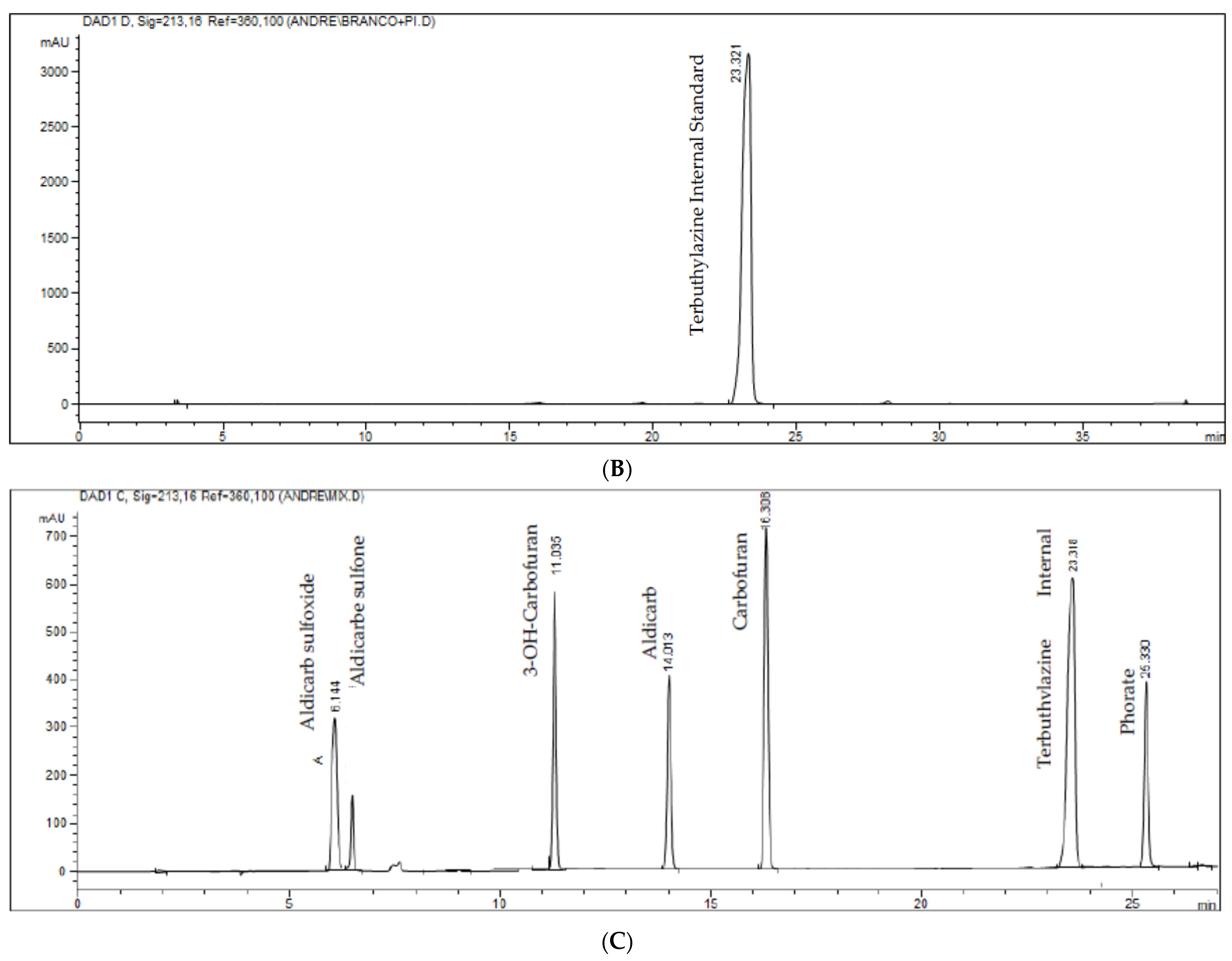
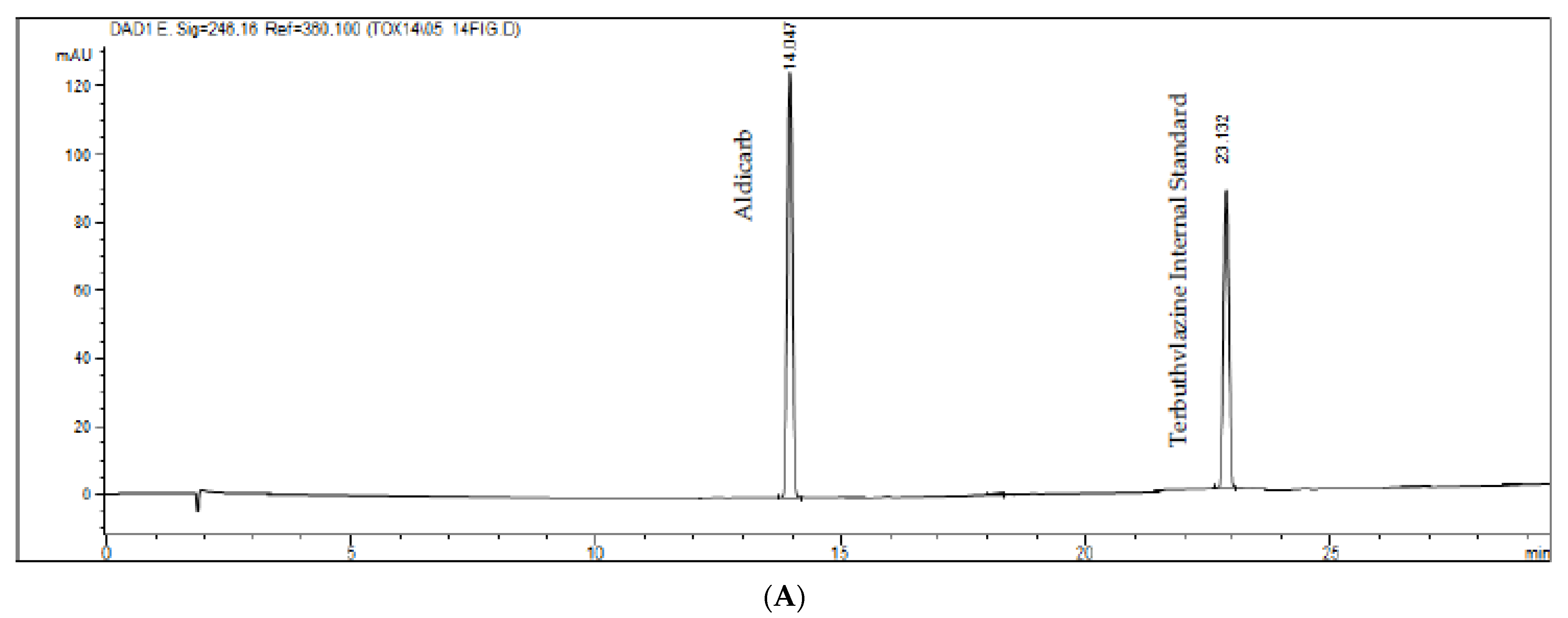
Figure 3.
(A–C). Chromatograms of white samples represented in figure (A), zero (white sample added to the internal standard) represented in figure (B) and standards (aldicarb–sulfoxide, aldicarb–sulfone, aldicarb, 3-OH-cabrofuran, carbofuran, internal standard and phorate) represented in figure (C). The pattern was added to a pool of all matrices evaluated.
Figure 3.
(A–C). Chromatograms of white samples represented in figure (A), zero (white sample added to the internal standard) represented in figure (B) and standards (aldicarb–sulfoxide, aldicarb–sulfone, aldicarb, 3-OH-cabrofuran, carbofuran, internal standard and phorate) represented in figure (C). The pattern was added to a pool of all matrices evaluated.
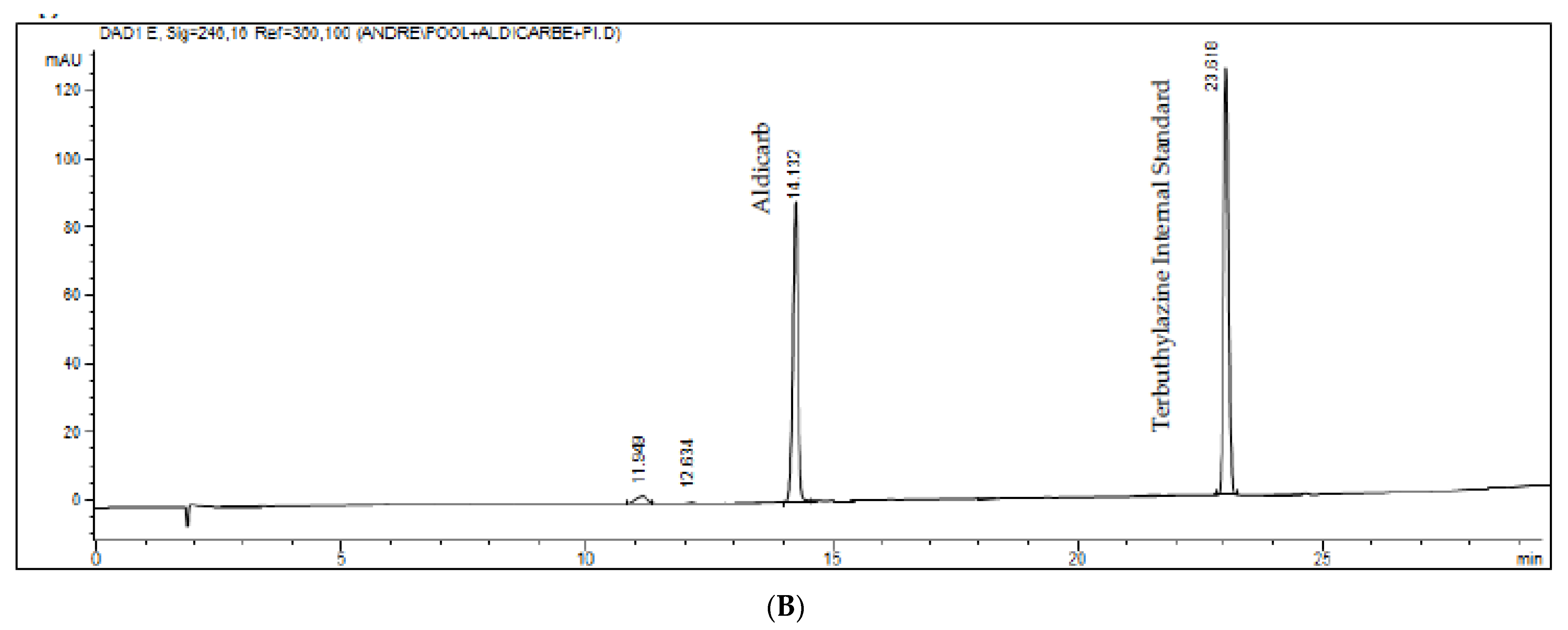
Figure 4.
(A,B). Chromatograms of the white sample added from the internal standard and the aldicarb figure (A). Standard was added in a pool of all the matrices evaluated and the actual sample figure (B).
Figure 4.
(A,B). Chromatograms of the white sample added from the internal standard and the aldicarb figure (A). Standard was added in a pool of all the matrices evaluated and the actual sample figure (B).
Table 1.
Commercially available carbamates and organophosphates, adapted from [
26].
Table 1.
Commercially available carbamates and organophosphates, adapted from [
26].
| Carbamates | Organophosphates |
|---|
| Aldicarb | Acephate | Menazon |
| Aminocarb | Acetion | Merfos |
| Carbaryl | Cyanophos | Methamidophos |
| Carbofuran | Chlorthion | Ronel |
| Landrin | Crufomate | Temephos |
| Metacalmate | Phenithrotion | Tetrachlorvinphos |
| Methiocarb | Formotion | Propyl Thiopyrophosphate |
| Mexacarbate | Fostex | Tribufun |
| Propoxur | Iodofenphos | Trichlorfon |
| | Malathion | |
Table 2.
Precision in each matrix for aldicarb was evaluated by means of the coefficient of variation (CV%) of lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
Table 2.
Precision in each matrix for aldicarb was evaluated by means of the coefficient of variation (CV%) of lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 8.1 | 5.0 | 3.7 | 3.2 |
| Stomach contents | 5.9 | 5.5 | 8.5 | 0.6 |
| Brain | 4.3 | 5.1 | 5.4 | 3.4 |
| Heart | 15.0 | 20.0 | 15.3 | 13.9 |
| Liver | 10.2 | 3.9 | 8.6 | 1.5 |
| Vitreous humor | 9.3 | 5.6 | 4.5 | 4.6 |
| Lung | 4.0 | 4.5 | 8.6 | 4.2 |
| Kidneys | 6.0 | 4.7 | 7.5 | 5.6 |
Table 3.
Variant coefficient of the calibration curves for aldicarb, aldicarb–sulfoxide, aldicarb–sulfone, carbofuran, and 3-OH-carbofuran for the analyzed matrices.
Table 3.
Variant coefficient of the calibration curves for aldicarb, aldicarb–sulfoxide, aldicarb–sulfone, carbofuran, and 3-OH-carbofuran for the analyzed matrices.
| Matrices | Coefficient of Variation |
|---|
| Aldicarb | Aldicarb–Sulfoxide | Aldicarb–Sulfone | Carbofuran | 3-OH-Carbofuran |
|---|
| Standard | 0.12% | 0.12% | 0.15% | 0.15% | 0.20% |
| Blood | 4.66% | 3.65% | 4.67% | 2.53% | 2.65% |
| Stomach contents | 5.18% | 2.16% | 4.15% | 2.07% | 2.53% |
| Brain | 4.70% | 5.73% | 3.72% | 3.18% | 4.10% |
| Heart | 19.68% | 18.38% | 16.98% | 2.81% | 2.03% |
| Liver | 7.28% | 4.26% | 6.48% | 1.50% | 3.35% |
| Vitreous humor | 5.08% | 2.18% | 1.08% | 1.22% | 1.92% |
| Lung | 4.57% | 4.57% | 3.27% | 4.57% | 8.68% |
| Kidneys | 5.99% | 4.99% | 2.43% | 5.99% | 10.42% |
Table 4.
Precision in each matrix for aldicarb–sulfoxide was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
Table 4.
Precision in each matrix for aldicarb–sulfoxide was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 8.2 | 5.0 | 3.2 | 3.1 |
| Stomach contents | 6.0 | 5.1 | 8.1 | 0.4 |
| Brain | 4.1 | 5.4 | 5.2 | 3.1 |
| Heart | 14.3 | 19.1 | 15.1 | 13.0 |
| Liver | 10.1 | 4.1 | 8.4 | 1.1 |
| Vitreous humor | 9.4 | 5.2 | 4.2 | 4.2 |
| Lung | 4.1 | 4.3 | 8.3 | 4.1 |
| Kidneys | 6.2 | 4.5 | 7.3 | 5.1 |
Table 5.
Precision in each matrix for aldicarb–sulfone was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
Table 5.
Precision in each matrix for aldicarb–sulfone was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 7.2 | 5.1 | 3.1 | 3.2 |
| Stomach contents | 4.0 | 5.2 | 8.0 | 0.5 |
| Brain | 4.4 | 5.1 | 5.0 | 3.2 |
| Heart | 12.3 | 16.1 | 14.1 | 10.0 |
| Liver | 9.1 | 4.2 | 8.5 | 1.1 |
| Vitreous humor | 8.2 | 5.1 | 4.1 | 4.1 |
| Lung | 4.5 | 4.5 | 8.1 | 4.4 |
| Kidneys | 4.2 | 4.1 | 7.6 | 5.2 |
Table 6.
Precision in each matrix for carbofuran was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
Table 6.
Precision in each matrix for carbofuran was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 7.2 | 4.1 | 1.1 | 1.5 |
| Stomach contents | 4.3 | 1.2 | 5.0 | 0.7 |
| Brain | 4.1 | 5.1 | 2.0 | 1.4 |
| Heart | 11.3 | 12.1 | 11.1 | 11.0 |
| Liver | 6.1 | 3.2 | 8.1 | 2.6 |
| Vitreous humor | 8.4 | 4.1 | 4.2 | 6.2 |
| Lung | 1.5 | 1.5 | 4.1 | 2.1 |
| Kidneys | 2.2 | 6.1 | 1.6 | 4.2 |
Table 7.
Precision in each matrix for 3-OH- carbofuran was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
Table 7.
Precision in each matrix for 3-OH- carbofuran was evaluated by means of the coefficient of variation (CV%) of the lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC).
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 2.2 | 2.1 | 1.2 | 2.5 |
| Stomach contents | 6.3 | 3.2 | 3.1 | 0.4 |
| Brain | 2.1 | 3.1 | 2.4 | 1.2 |
| Heart | 10.1 | 11.5 | 4.1 | 9.0 |
| Liver | 3.5 | 4.2 | 3.1 | 3.6 |
| Vitreous humor | 3.4 | 2.1 | 2.2 | 3.2 |
| Lung | 3.5 | 2.5 | 2.1 | 2.4 |
| Kidneys | 2.5 | 3.5 | 1.5 | 2.2 |
Table 8.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to aldicarb.
Table 8.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to aldicarb.
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 6.97 | 2.16 | 11.34 | 3.2 |
| Stomach contents | 1.82 | 5.77 | 10.58 | 8.72 |
| Brain | 11.53 | 4.28 | 0.67 | 1.27 |
| Heart | 14.09 | 11.59 | 13.94 | 11.27 |
| Liver | 12.13 | 17.36 | 9.89 | 3.31 |
| Vitreous humor | 11.69 | 9.6 | 3.74 | 1.55 |
| Lung | 7.32 | 6.47 | 6.28 | 9.14 |
| Kidneys | 10.2 | 19.32 | 10.34 | 2.44 |
Table 9.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to aldicarb–sulfoxide.
Table 9.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to aldicarb–sulfoxide.
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 5.63 | 1.14 | 12.32 | 2.22 |
| Stomach contents | 2.01 | 4.72 | 9.52 | 6.23 |
| Brain | 12.01 | 3.13 | 0.99 | 1.67 |
| Heart | 12.09 | 10.01 | 10.12 | 10.22 |
| Liver | 12.90 | 14.22 | 8.22 | 1.2 |
| Vitreous humor | 10.45 | 8.12 | 4.23 | 1.65 |
| Lung | 5.31 | 4.23 | 5.21 | 8.23 |
| Kidneys | 9.2 | 14.12 | 9.32 | 1.22 |
Table 10.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to aldicarb–sulfone.
Table 10.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to aldicarb–sulfone.
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 4.22 | 2.41 | 10.25 | 1.55 |
| Stomach contents | 1.29 | 3.91 | 8.12 | 4.11 |
| Brain | 11.11 | 2.23 | 1.00 | 1.11 |
| Heart | 11.19 | 9.34 | 9.09 | 9.1 |
| Liver | 13.91 | 13.31 | 7.55 | 2.21 |
| Vitreous humor | 11.66 | 7.33 | 3.12 | 1.43 |
| Lung | 3.23 | 2.12 | 4.24 | 7.21 |
| Kidneys | 10.12 | 10.22 | 10.12 | 1.55 |
Table 11.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control, (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to carbofuran.
Table 11.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control, (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to carbofuran.
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 5.67 | 3.22 | 9.45 | 2.87 |
| Stomach contents | 3.02 | 2.51 | 7.54 | 3.13 |
| Brain | 10.02 | 1.22 | 2.00 | 2.53 |
| Heart | 10.91 | 9.04 | 9.01 | 9.15 |
| Liver | 12.91 | 12.71 | 8.59 | 3.26 |
| Vitreous humor | 10.65 | 4.32 | 2.54 | 2.22 |
| Lung | 2.43 | 1.23 | 2.12 | 5.24 |
| Kidneys | 9.11 | 9.23 | 9.27 | 1.05 |
Table 12.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to 3-OH-carbofuran.
Table 12.
Lower limit of quantification (LLQ), low-quality control (LQC), medium-quality control (MQC), and high-quality control (HQC) calculated in each matrix, in triplicate, intracutaneous and intercurrent to 3-OH-carbofuran.
| Biological Matrices | LLQ% | LQC% | MQC% | HQC% |
|---|
| Blood | 4.89 | 3.01 | 8.12 | 3.44 |
| Stomach contents | 2.11 | 2.32 | 6.23 | 3.08 |
| Brain | 9.01 | 1.76 | 1.65 | 1.99 |
| Heart | 9.08 | 10.23 | 10.01 | 10.11 |
| Liver | 11.23 | 11.12 | 7.29 | 1.24 |
| Vitreous humor | 9.35 | 3.33 | 1.66 | 1.99 |
| Lung | 1.42 | 2.31 | 3.54 | 4.22 |
| Kidneys | 8.12 | 7.53 | 8.17 | 0.92 |
Table 13.
Presentation of mean recovery data in % of all analytes and different biological matrices [
54].
Table 13.
Presentation of mean recovery data in % of all analytes and different biological matrices [
54].
| Biological Matrices | Average Recovery of Adicarb | Average Recovery of Adicarb–Sulfoxide | Average Recovery of Adicarb Sulfone | Average Recovery of Carbofuran | Average Recovery of 3-OH-Carbofuran |
|---|
| Blood | 60% | 62% | 61% | 60% | 64% |
| Stomach contents | 70% | 73% | 72% | 69% | 70% |
| Brain | 46% | 50% | 43% | 47% | 44% |
| Heart | 30% | 29% | 29% | 31% | 33% |
| Liver | 57% | 66% | 60% | 53% | 60% |
| Vitreous humor | 32% | 43% | 40% | 31 | 44% |
| Lung | 31% | 30% | 32% | 33% | 29% |
| Kidneys | 48% | 32% | 33% | 50% | 31% |
Table 14.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of aldicarb–sulfone.
Table 14.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of aldicarb–sulfone.
| Biological Matrices | CV LLQ | CV Medium | CV Medium ISD | CV SLQ | CV Medium | CV Medium ISD |
|---|
| Blood | 8.1% | 4.7% | 5.7% | 3.8% | 6.7% | 2.4% |
| Stomach contents | 5.9% | 5.2% | 2.6% | 4.6% | 8.1% | 2.3% |
| Brain | 4.3% | 4.7% | 7.8% | 3.7% | 3.0% | 2.2% |
| Heart | 15% | 10.2% | 11.7% | 12.7% | 11.6% | 9.6% |
| Liver | 10.2% | 7.1% | 11.5% | 8.8% | 8.0% | 1.7% |
| Vitreous humor | 9.3% | 5.0% | 9.9% | 6.2% | 3.6% | 2.8% |
| Lung | 4.0% | 4.5% | 3.9% | 3.3% | 6.2% | 5.8% |
| Kidneys | 6.0% | 5.9% | 7.2% | 7.2% | 8.9% | 3.4% |
Table 15.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of 3-OH-carbofuran.
Table 15.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of 3-OH-carbofuran.
| Biological Matrices | CV LLQ | CV Medium | CV Medium ISD | CV SLQ | CV Medium | CV Medium ISD |
|---|
| Blood | 4.1% | 4.7% | 2.0% | 2.2% | 6.7% | 6.0% |
| Stomach contents | 2.1% | 5.2% | 3.0% | 5.7% | 4.2% | 5.0% |
| Brain | 3.1% | 4.7% | 2.0% | 4.2% | 1.7% | 3.0% |
| Heart | 7.0% | 10.2% | 3.0% | 12.2% | 2.2% | 2.0% |
| Liver | 5.2% | 7.1% | 4.0% | 5.3% | 5.1% | 1.0% |
| Vitreous humor | 9.3% | 5.0% | 6.0% | 2.5% | 2.0% | 4.0% |
| Lung | 4.0% | 4.5% | 2.0% | 5.1% | 2.5% | 2.0% |
| Kidneys | 6.0% | 5.9% | 1.0% | 3.4% | 3.9% | 2.0% |
Table 16.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of aldicarb.
Table 16.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of aldicarb.
| Biological Matrices | CV LLQ | CV Medium | CV Medium ISD | CV SLQ | CV Medium | CV Medium ISD |
|---|
| Blood | 7.5% | 3.6% | 7.5% | 3.2% | 8.1% | 4.7% |
| Stomach contents | 3.9% | 5.6% | 9.5% | 4.7% | 5.9% | 5.2% |
| Brain | 7.9% | 4.7% | 3.0% | 2.3% | 4.3% | 4.7% |
| Heart | 14.5% | 15.8% | 14.6% | 12.6% | 15.0% | 10.2% |
| Liver | 11.2% | 10.6% | 9.2% | 2.4% | 10.2% | 7.1% |
| Vitreous humor | 10.5% | 7.6% | 4.1% | 3.1% | 9.3% | 5.0% |
| Lung | 5.7% | 5.5% | 7.4% | 6.7% | 4.0% | 4.5% |
| Kidneys | 8.1% | 12.0% | 8.9% | 4.0% | 6.0% | 5.9% |
Table 17.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of aldicarb–sulfoxide.
Table 17.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of aldicarb–sulfoxide.
| Biological Matrices | CV LLQ | CV Medium | CV Medium ISD | CV SLQ | CV Medium | CV Medium ISD |
|---|
| Blood | 7.2% | 3.3% | 3.4% | 3.1% | 7.2% | 2.2% |
| Stomach contents | 6.0% | 4.1% | 4.8% | 7.6% | 4.9% | 5.1% |
| Brain | 2.1% | 3.4% | 3.1% | 2.4% | 2.1% | 2.1% |
| Heart | 15.0% | 10.2% | 4.0% | 11.2% | 15.0% | 10.2% |
| Liver | 10.2% | 7.1% | 4.0% | 3.3% | 10.2% | 7.1% |
| Vitreous humor | 9.3% | 5.0% | 5.0% | 1.5% | 9.3% | 5.0% |
| Lung | 4.0% | 4.5% | 4.0% | 9.1% | 4.0% | 4.5% |
| Kidneys | 6.0% | 5.9% | 5.0% | 2.4% | 6.0% | 5.9% |
Table 18.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of carbofuran.
Table 18.
Coefficient of variation (CV) of the lower limit of quantification (LLQ) and the superior limit of quantification (SLQ), as well as the average CV (average of the CV of the other points of the calibration curve), and of the internal standard (ISD) in the different matrices, expressed in percentage of carbofuran.
| Biological Matrices | CV LLQ | CV Medium | CV Medium ISD | CV SLQ | CV Medium | CV Medium ISD |
|---|
| Blood | 4.0% | 6.4% | 3.7% | 5.3% | 2.2% | 3.2% |
| Stomach contents | 5.0% | 3.7% | 1.9% | 6.3% | 1.9% | 8.7% |
| Brain | 4.0% | 7.1% | 3.2% | 2.0% | 2.0% | 1.2% |
| Heart | 4.0% | 11.1% | 10.6% | 10.1% | 10.1% | 11.2% |
| Liver | 4.0% | 9.5% | 8.0% | 8.3% | 2.9% | 3.3% |
| Vitreous humor | 5.0% | 9.5% | 4.2% | 3.4% | 4.2% | 1.5% |
| Lung | 4.0% | 2.0% | 1.4% | 3.1% | 3.7% | 9.1% |
| Kidneys | 5.0% | 5.7% | 7.7% | 5.4% | 2.6% | 2.4% |
Table 19.
Cases of suspected poisoning by carbamate pesticides (aldicarb and carbofuran) in animals whose tissue and feed samples were sent to the Laboratory of Toxicological Diagnosis (LADTOX) from 2010 to June 2015.
Table 19.
Cases of suspected poisoning by carbamate pesticides (aldicarb and carbofuran) in animals whose tissue and feed samples were sent to the Laboratory of Toxicological Diagnosis (LADTOX) from 2010 to June 2015.
| Samples | | | Years | | | |
|---|
| 2010 | 2011 | 2012 | 2013 | 2014 | 2015 |
|---|
| Dogs | 12 | 11 | 11 | 18 | 07 | 07 |
| Positive | 07 | 07 | 08 | 12 | 06 | 04 |
| Negative | 04 | 11 | 03 | 06 | 01 | 03 |
| Cats | 05 | 25 | 11 | 22 | 19 | 03 |
| Positive | 05 | 22 | 06 | 16 | 14 | 02 |
| Negative | | 03 | 05 | 06 | 05 | 01 |
| Ration | | 01 | 01 | | 01 | |
| Positive | | 01 | | | 01 | |
| Negative | | | 01 | | | |
| Birds | | 08 | | | | |
| Positive | | 08 | | | | |
| Negative | | | | | | |
| Cattle | 01 | | 01 | | 01 | 01 |
| Positive | | | | | 01 | |
| Negative | 01 | | 01 | | | 01 |
| Others | | 01 a | 01 b | | | |
| Positive | | 01 | 01 | | | |
| Negative | | | - | | | |
| Total | 18 | 48 | 26 | 40 | 28 | 12 |
| Positive | 12 | 42 | 14 | 28 | 23 | 06 |
| Negative | 06 | 06 | 12 | 12 | 05 | 06 |
Table 20.
Cases of suspected poisoning by the pesticide aldicarb in animals whose tissue and feed samples were sent to the Laboratory of Toxicological Diagnosis (LADTOX) from 2014 to June 2015.
Table 20.
Cases of suspected poisoning by the pesticide aldicarb in animals whose tissue and feed samples were sent to the Laboratory of Toxicological Diagnosis (LADTOX) from 2014 to June 2015.
| Material | Stomach Contents | Liver | Blood | Ration | Species |
|---|
| 1 | - | + | - | + | Dog |
| 2 | - | - | - | - | Cat |
| 3 | - | + | - | - | Cat |
| 4 | - | - | - | - | Cat |
| 5 | - | - | - | - | Dog |
| 6 | + | + | - | - | Cat |
| 7 | - | - | + | - | Dog |
| 8 | + | - | - | - | Cat |
| 9 | - | - | - | - | Dog |
| 10 | + | - | - | - | Cat |
| 11 | - | - | - | - | Dog |
| 12 | + | + | - | - | Cat |
| 13 | + | + | - | - | Cat |
| 15 | + | | - | - | Cat |
| 16 | | | - | + | Dog |
| 17 | - | - | - | - | Cat |
| 18 | + | + | - | - | Dog |
| 19 | - | - | - | - | Cat |
| 20 | + | + | - | - | Cat |
| 21 | - | - | - | - | Cat |
| 22 | + | + | - | - | Cat |
| 23 | + | - | - | - | Cattle |
| 24 | - | - | - | - | Cat |
| 25 | + | - | - | - | Cat |
| 26 | + | - | - | - | Cat |
| 27 | + | - | - | - | Cat |
| 28 | + | - | - | - | Cat |
| 29 | + | - | - | - | Dog |
| 30 | + | - | - | - | Skunk |
| 31 | - | - | - | - | Dog |
| 32 | - | - | - | - | Dog |
| 33 | - | - | - | - | Cat |
| 34 | + | + | - | - | Dog |
| 35 | + | - | - | - | Dog |
| 36 | - | - | - | - | Dog |
| 37 | - | - | - | - | Dog |
| 38 | - | - | - | - | Dog |
| 39 | - | - | - | - | Cattle |
| 40 | - | + | - | - | Cat |
| 41 | - | - | - | - | Dog |
| 42 | - | - | - | - | Dog |
| 43 | - | - | - | - | Cat |
| 44 | - | - | - | - | Cat |
| 45 | + | - | - | + | Dog |
| 46 | - | - | - | - | Cat |
| 47 | - | - | - | - | Cat |
| 48 | - | - | - | - | Chicken |
| 49 | - | - | - | - | Chicken |
| 50 | - | - | - | - | Chicken |
| 51 | - | - | - | - | Dog |
| Positive | 19 | 10 | 01 | 03 | |
| Negative | 25 | 26 | 02 | 0 | |
| Total | 44 | 36 | 03 | 03 | |


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}