
Epigenetics and Methylmercury-Induced Neurotoxicity, Evidence from Experimental Studies

 , ,
, , 
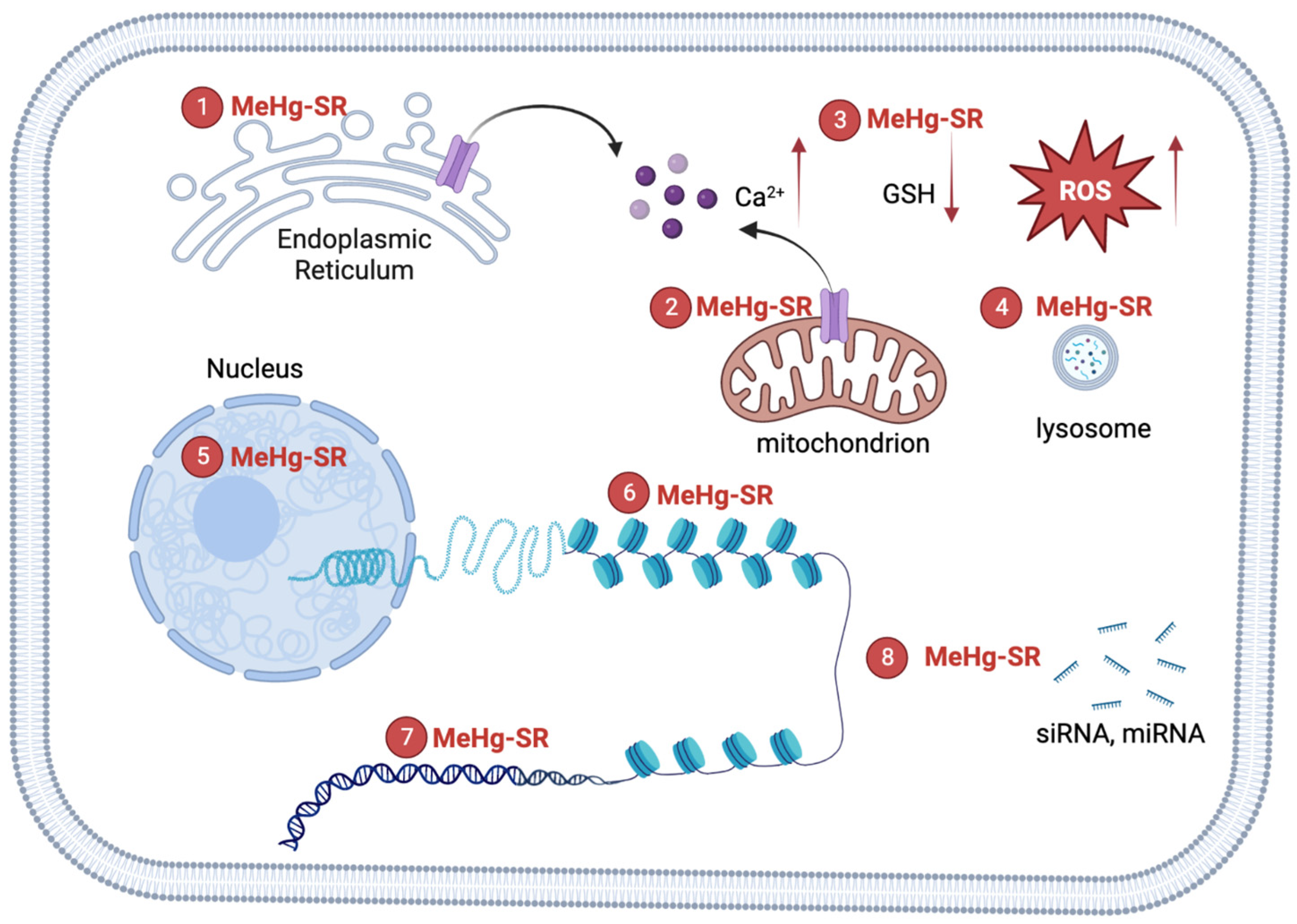{kind=link}
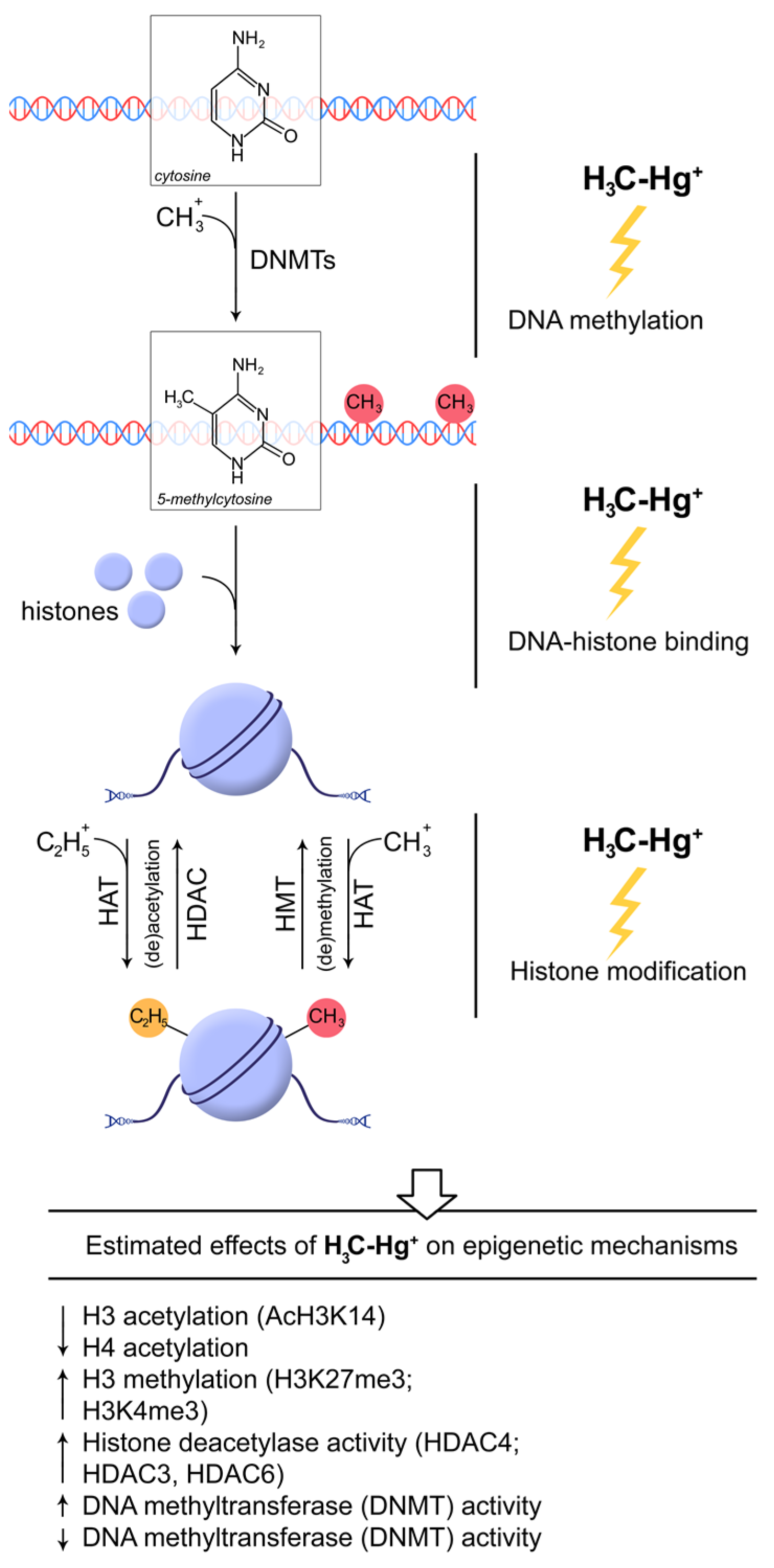{kind=link}
Abstract
:1. Introduction
2. MeHg, DNA, and Chromatin
3. MeHg and Neurogenesis
4. MeHg, miRNA, and RNA Interference (RNAi)
5. Transgenerational Neurotoxicity of MeHg
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cediel Ulloa, A.; Gliga, A.; Love, T.M.; Pineda, D.; Mruzek, D.W.; Watson, G.E.; Davidson, P.W.; Shamlaye, C.F.; Strain, J.J.; Myers, G.J.; et al. Prenatal methylmercury exposure and DNA methylation in seven-year-old children in the Seychelles Child Development Study. Environ. Int. 2021, 147, 106321. [Google Scholar] [CrossRef]
- Nogara, P.A.; Oliveira, C.S.; Schmitz, G.L.; Piquini, P.C.; Farina, M.; Aschner, M.; Rocha, J.B.T. Methylmercury’s chemistry: From the environment to the mammalian brain. Biochim. Biophys. Acta BBA Gen. Subj. 2019, 1863, 129284. [Google Scholar] [CrossRef]
- Barone, G.; Storelli, A.; Meleleo, D.; Dambrosio, A.; Garofalo, R.; Busco, A.; Storelli, M.M. Levels of Mercury, Methylmercury and Selenium in Fish: Insights into Children Food Safety. Toxics 2021, 9, 39. [Google Scholar] [CrossRef]
- Ortega-García, J.A.; Rodriguez, K.; Calatayud, M.; Martin, M.; Vélez, D.; Devesa, V.; Sánchez-Alarcon, M.C.; Torres Cantero, A.M.; Galindo-Cascales, C.; Gil-Vázquez, J.M.; et al. Estimated intake levels of methylmercury in children, childbearing age and pregnant women in a Mediterranean region, Murcia, Spain. Eur. J. Pediatr. 2009, 168, 1075–1080. [Google Scholar] [CrossRef]
- Mason, R.P. The Bioaccumulation of Mercury, Methylmercury, and Other Toxic Elements into Pelagic and Benthic Organisms. In Coastal and Estuarine Risk Assessment; CRC Press: Boca Raton, FL, USA, 2001; pp. 143–166. [Google Scholar]
- Raes, B.B. The ultrastructural effect and subcellular localization of mercuric chloride and methylmercuric chloride in insect cells (Aedes albopictus C6/36). Tissue Cell 1999, 31, 223–232. [Google Scholar] [CrossRef]
- Komsta-Szumska, E.; Reuhl, K.R.; Miller, D.R. The effect of methylmercury on the distribution and excretion of selenium by the guinea pig. Arch. Toxicol. 1983, 54, 303–310. [Google Scholar] [CrossRef]
- Eto, K. Pathology of Minamata disease. Toxicol. Pathol. 1997, 25, 614–623. [Google Scholar] [CrossRef] [Green Version]
- Farina, M.; Aschner, M. Glutathione antioxidant system and methylmercury-induced neurotoxicity: An intriguing interplay. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129285. [Google Scholar] [CrossRef]
- Yuan, Y.; Atchison, W.D. Multiple Sources of Ca2+ Contribute to Methylmercury-Induced Increased Frequency of Spontaneous Inhibitory Synaptic Responses in Cerebellar Slices of Rat. Toxicol. Sci. 2016, 150, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Ishida, Y.; Takahashi, T.; Naganuma, A.; Hwang, G.W. Transport of pyruvate into mitochondria is involved in methylmercury toxicity. Sci. Rep. 2016, 6, 21528. [Google Scholar] [CrossRef]
- Ke, T.; Rocha, J.B.T.; Tinkov, A.A.; Santamaria, A.; Bowman, A.B.; Aschner, M. The Role of Human LRRK2 in Acute Methylmercury Toxicity in Caenorhabditis elegans. Neurochem. Res. 2021, 46, 2991–3002. [Google Scholar] [CrossRef]
- Usuki, F.; Fujimura, M.; Yamashita, A. Endoplasmic reticulum stress preconditioning modifies intracellular mercury content by upregulating membrane transporters. Sci. Rep. 2017, 7, 12390. [Google Scholar] [CrossRef] [Green Version]
- Takanezawa, Y.; Nakamura, R.; Hamaguchi, M.; Yamamoto, K.; Sone, Y.; Uraguchi, S.; Kiyono, M. Docosahexaenoic acid enhances methylmercury-induced endoplasmic reticulum stress and cell death and eicosapentaenoic acid potentially attenuates these effects in mouse embryonic fibroblasts. Toxicol. Lett. 2019, 306, 35–42. [Google Scholar] [CrossRef]
- Choi, B.H.; Simpkins, H. Changes in the molecular structure of mouse fetal astrocyte nucleosomes produced in vitro by methylmercuric chloride. Env. Res. 1986, 39, 321–330. [Google Scholar] [CrossRef]
- Maki, A.H.; Ott, C.M. Methylmercury(II) binding to single-stranded and duplex DNA: Complexes formed are distinguishable by optical detection of magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 1981, 78, 2972–2976. [Google Scholar] [CrossRef] [Green Version]
- Carvan, M.J., 3rd; Kalluvila, T.A.; Klingler, R.H.; Larson, J.K.; Pickens, M.; Mora-Zamorano, F.X.; Connaughton, V.P.; Sadler-Riggleman, I.; Beck, D.; Skinner, M.K. Mercury-induced epigenetic transgenerational inheritance of abnormal neurobehavior is correlated with sperm epimutations in zebrafish. PLoS ONE 2017, 12, e0176155. [Google Scholar] [CrossRef] [Green Version]
- Onishchenko, N.; Karpova, N.; Sabri, F.; Castrén, E.; Ceccatelli, S. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J. Neurochem. 2008, 106, 1378–1387. [Google Scholar] [CrossRef]
- Bose, R.; Onishchenko, N.; Edoff, K.; Janson Lang, A.M.; Ceccatelli, S. Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicol. Sci. 2012, 130, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Onyido, I.; Norris, A.R.; Buncel, E. Biomolecule-mercury interactions: Modalities of DNA base-mercury binding mechanisms. Remediation strategies. Chem. Rev. 2004, 104, 5911–5929. [Google Scholar] [CrossRef]
- Beerman, T.A.; Lebowitz, J. Further analysis of the altered secondary structure of superhelical DNA. Sensitivity to methylmercuric hydroxide a chemical probe for unpaired bases. J. Mol. Biol. 1973, 79, 451–470. [Google Scholar] [CrossRef]
- Bailey, J.M.; Davidson, N. Methylmercury as a reversible denaturing agent for agarose gel electrophoresis. Anal. Biochem. 1976, 70, 75–85. [Google Scholar] [CrossRef]
- Vicari, T.; Ferraro, M.V.; Ramsdorf, W.A.; Mela, M.; de Oliveira Ribeiro, C.A.; Cestari, M.M. Genotoxic evaluation of different doses of methylmercury (CH₃Hg⁺) in Hoplias malabaricus. Ecotoxicol. Environ. Saf. 2012, 82, 47–55. [Google Scholar] [CrossRef]
- Ondovcik, S.L.; Tamblyn, L.; McPherson, J.P.; Wells, P.G. Oxoguanine glycosylase 1 (OGG1) protects cells from DNA double-strand break damage following methylmercury (MeHg) exposure. Toxicol. Sci. 2012, 128, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Lerebours, A.; Cambier, S.; Hislop, L.; Adam-Guillermin, C.; Bourdineaud, J.P. Genotoxic effects of exposure to waterborne uranium, dietary methylmercury and hyperoxia in zebrafish assessed by the quantitative RAPD-PCR method. Mutat. Res. 2013, 755, 55–60. [Google Scholar] [CrossRef]
- Crespo-Lopez, M.E.; Costa-Malaquias, A.; Oliveira, E.H.; Miranda, M.S.; Arrifano, G.P.; Souza-Monteiro, J.R.; Sagica, F.E.; Fontes-Junior, E.A.; Maia, C.S.; Macchi, B.M.; et al. Is Low Non-Lethal Concentration of Methylmercury Really Safe? A Report on Genotoxicity with Delayed Cell Proliferation. PLoS ONE 2016, 11, e0162822. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.H.; Pereira, J.P.G.; Malaquias, A.C.; Sagica, F.; de Oliveira, E.H.C. Intracellular accumulation and DNA damage caused by methylmercury in glial cells. J. Biochem. Mol. Toxicol. 2022, 36, e23170. [Google Scholar] [CrossRef]
- Yin, Z.; Jiang, H.; Syversen, T.; Rocha, J.B.; Farina, M.; Aschner, M. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J. Neurochem. 2008, 107, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Rabenstein, D.L. Chemistry of methylmercury toxicology. J. Chem. Educ. 1978, 55, 292. [Google Scholar] [CrossRef]
- Patnaik, R.; Padhy, R.N. Comparative study on toxicity of methylmercury chloride and methylmercury hydroxide to the human neuroblastoma cell line SH-SY5Y. Environ. Sci. Pollut. Res. Int. 2018, 25, 20606–20614. [Google Scholar] [CrossRef]
- Lemes, M.; Wang, F. Methylmercury speciation in fish muscle by HPLC-ICP-MS following enzymatic hydrolysis. J. Anal. At. Spectrom. 2009, 24, 663–668. [Google Scholar] [CrossRef]
- Chanda, S.K.; Cherian, M.G. Isolation and partial characterization of a mercury-binding nonhistone protein component from rat kidney nuclei. Biochem. Biophys. Res. Commun. 1973, 50, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Gruenwedel, D.W.; Glaser, J.F.; Cruikshank, M.K. Binding of methylmercury(II) by HeLa S3 suspension-culture cells: Intracellular methylmercury levels and their effect on DNA replication and protein synthesis. Chem. Biol. Interact. 1981, 36, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, L.G.; Gruenwedel, D.W. Methylmercury-chromosome interactions. I. Thermal denaturation of calf thymus chromatin in presence of CH3HgOH. Z Nat. C Biosci. 1980, 35, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.M.; Henikoff, S. Histone variants: Dynamic punctuation in transcription. Genes Dev. 2014, 28, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.S.; Shannon, M.F.; Hardy, K. Epigenetic control of inducible gene expression in the immune system. Epigenomics 2010, 2, 775–795. [Google Scholar] [CrossRef]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef]
- Rudgalvyte, M.; VanDuyn, N.; Aarnio, V.; Heikkinen, L.; Peltonen, J.; Lakso, M.; Nass, R.; Wong, G. Methylmercury exposure increases lipocalin related (lpr) and decreases activated in blocked unfolded protein response (abu) genes and specific miRNAs in Caenorhabditis elegans. Toxicol. Lett. 2013, 222, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Prince, L.M.; Rand, M.D. Notch Target Gene E(spl)mδ Is a Mediator of Methylmercury-Induced Myotoxicity in Drosophila. Front. Genet. 2017, 8, 233. [Google Scholar] [CrossRef]
- Gunderson, J.T.; Peppriell, A.E.; Vorojeikina, D.; Rand, M.D. Tissue-specific Nrf2 signaling protects against methylmercury toxicity in Drosophila neuromuscular development. Arch. Toxicol. 2020, 94, 4007–4022. [Google Scholar] [CrossRef] [PubMed]
- Diana Neely, M.; Xie, S.; Prince, L.M.; Kim, H.; Tukker, A.M.; Aschner, M.; Thimmapuram, J.; Bowman, A.B. Single cell RNA sequencing detects persistent cell type- and methylmercury exposure paradigm-specific effects in a human cortical neurodevelopmental model. Food Chem. Toxicol. 2021, 154, 112288. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Xu, Y.; Xu, S.; Cheng, L.; Zhou, T.; Xie, A.; Xu, A.; Wu, L.; Chen, S. Ecotoxicity Risk of Low-Dose Methylmercury Exposure to Caenorhabditis elegans: Multigenerational Toxicity and Population Discrepancy. Chem. Res. Toxicol. 2021, 34, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Prince, L.M.; Neely, M.D.; Warren, E.B.; Thomas, M.G.; Henley, M.R.; Smith, K.K.; Aschner, M.; Bowman, A.B. Environmentally relevant developmental methylmercury exposures alter neuronal differentiation in a human-induced pluripotent stem cell model. Food Chem. Toxicol. 2021, 152, 112178. [Google Scholar] [CrossRef]
- Waterhouse, E.G.; An, J.J.; Orefice, L.L.; Baydyuk, M.; Liao, G.Y.; Zheng, K.; Lu, B.; Xu, B. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J. Neurosci. 2012, 32, 14318–14330. [Google Scholar] [CrossRef] [Green Version]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Rossi, C.; Angelucci, A.; Costantin, L.; Braschi, C.; Mazzantini, M.; Babbini, F.; Fabbri, M.E.; Tessarollo, L.; Maffei, L.; Berardi, N.; et al. Brain-derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur. J. Neurosci. 2006, 24, 1850–1856. [Google Scholar] [CrossRef]
- Kuzumaki, N.; Ikegami, D.; Tamura, R.; Hareyama, N.; Imai, S.; Narita, M.; Torigoe, K.; Niikura, K.; Takeshima, H.; Ando, T.; et al. Hippocampal epigenetic modification at the brain-derived neurotrophic factor gene induced by an enriched environment. Hippocampus 2011, 21, 127–132. [Google Scholar] [CrossRef]
- Lee, R.S.; Sawa, A. Environmental stressors and epigenetic control of the hypothalamic-pituitary-adrenal axis. Neuroendocrinology 2014, 100, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Desaulniers, D.; Xiao, G.H.; Cummings-Lorbetskie, C. Effects of lactational and/or in utero exposure to environmental contaminants on the glucocorticoid stress-response and DNA methylation of the glucocorticoid receptor promoter in male rats. Toxicology 2013, 308, 20–33. [Google Scholar] [CrossRef]
- Guida, N.; Laudati, G.; Anzilotti, S.; Sirabella, R.; Cuomo, O.; Brancaccio, P.; Santopaolo, M.; Galgani, M.; Montuori, P.; Di Renzo, G.; et al. Methylmercury upregulates RE-1 silencing transcription factor (REST) in SH-SY5Y cells and mouse cerebellum. Neurotoxicology 2016, 52, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, N.; Laudati, G.; Mascolo, L.; Valsecchi, V.; Sirabella, R.; Selleri, C.; Di Renzo, G.; Canzoniero, L.M.; Formisano, L. p38/Sp1/Sp4/HDAC4/BDNF Axis Is a Novel Molecular Pathway of the Neurotoxic Effect of the Methylmercury. Front. Neurosci. 2017, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. Lausanne 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, N.; Valsecchi, V.; Laudati, G.; Serani, A.; Mascolo, L.; Molinaro, P.; Montuori, P.; Di Renzo, G.; Canzoniero, L.M.; Formisano, L. The miR206-JunD Circuit Mediates the Neurotoxic Effect of Methylmercury in Cortical Neurons. Toxicol. Sci. 2018, 163, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Nerini-Molteni, S.; Mennecozzi, M.; Fabbri, M.; Sacco, M.G.; Vojnits, K.; Compagnoni, A.; Gribaldo, L.; Bremer-Hoffmann, S. MicroRNA profiling as a tool for pathway analysis in a human in vitro model for neural development. Curr. Med. Chem. 2012, 19, 6214–6223. [Google Scholar] [CrossRef]
- Miska, E.A.; Alvarez-Saavedra, E.; Abbott, A.L.; Lau, N.C.; Hellman, A.B.; McGonagle, S.M.; Bartel, D.P.; Ambros, V.R.; Horvitz, H.R. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007, 3, e215. [Google Scholar] [CrossRef] [Green Version]
- Dexheimer, P.J.; Wang, J.; Cochella, L. Two MicroRNAs Are Sufficient for Embryonic Patterning in C. elegans. Curr. Biol. 2020, 30, 5058–5065.e5. [Google Scholar] [CrossRef]
- Boehm, M.; Slack, F. A developmental timing microRNA and its target regulate life span in C. elegans. Science 2005, 310, 1954–1957. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Saavedra, E.; Horvitz, H.R. Many families of C. elegans microRNAs are not essential for development or viability. Curr. Biol. 2010, 20, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Massirer, K.B.; Perez, S.G.; Mondol, V.; Pasquinelli, A.E. The miR-35-41 family of microRNAs regulates RNAi sensitivity in Caenorhabditis elegans. PLoS Genet. 2012, 8, e1002536. [Google Scholar] [CrossRef]
- Simmer, F.; Tijsterman, M.; Parrish, S.; Koushika, S.P.; Nonet, M.L.; Fire, A.; Ahringer, J.; Plasterk, R.H. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr. Biol. 2002, 12, 1317–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, T.; Santamaria, A.; Farina, M.; Rocha, J.B.T.; Bowman, A.B.; Aschner, M. The Modulatory Role of sti-1 in Methylmercury-Induced Toxicity in Caenorhabditis elegans. Neurotox. Res. 2022, 40, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.; Crawford, N.; Martell, M.; Khalil, B.; Imtiaz, F.; Newell-Caito, J.L.; Caito, S. MicroRNA Expression Influences Methylmercury-Induced Lipid Accumulation and Mitochondrial Toxicity in Caenorhabditis elegans. Chem. Res. Toxicol. 2022, 35, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Billi, A.C.; Fischer, S.E.; Kim, J.K. Endogenous RNAi pathways in C. elegans. WormBook 2014, 1–49. [Google Scholar] [CrossRef]
- Gent, J.I.; Schvarzstein, M.; Villeneuve, A.M.; Gu, S.G.; Jantsch, V.; Fire, A.Z.; Baudrimont, A. A Caenorhabditis elegans RNA-directed RNA polymerase in sperm development and endogenous RNA interference. Genetics 2009, 183, 1297–1314. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Manoharan, A.P.; Harkins, T.T.; Bouffard, P.; Fitzpatrick, C.; Chu, D.S.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, J.K. 26G endo-siRNAs regulate spermatogenic and zygotic gene expression in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2009, 106, 18674–18679. [Google Scholar] [CrossRef] [Green Version]
- Gent, J.I.; Lamm, A.T.; Pavelec, D.M.; Maniar, J.M.; Parameswaran, P.; Tao, L.; Kennedy, S.; Fire, A.Z. Distinct phases of siRNA synthesis in an endogenous RNAi pathway in C. elegans soma. Mol. Cell 2010, 37, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Kumsta, C.; Hansen, M.C. elegans rrf-1 mutations maintain RNAi efficiency in the soma in addition to the germline. PLoS ONE 2012, 7, e35428. [Google Scholar] [CrossRef] [Green Version]
- Lehrbach, N.J.; Castro, C.; Murfitt, K.J.; Abreu-Goodger, C.; Griffin, J.L.; Miska, E.A. Post-developmental microRNA expression is required for normal physiology, and regulates aging in parallel to insulin/IGF-1 signaling in C. elegans. RNA 2012, 18, 2220–2235. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.G.; Pak, J.; Guang, S.; Maniar, J.M.; Kennedy, S.; Fire, A. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat. Genet. 2012, 44, 157–164. [Google Scholar] [CrossRef]
- Guang, S.; Bochner, A.F.; Burkhart, K.B.; Burton, N.; Pavelec, D.M.; Kennedy, S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 2010, 465, 1097–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, N.O.; Burkhart, K.B.; Kennedy, S. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2011, 108, 19683–19688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, M.; Aschner, M.; Rocha, J.B. Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharm. 2011, 256, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillard, P.V.; van der Veen, A.G.; Poirier, E.Z.; Reis e Sousa, C. Slicing and dicing viruses: Antiviral RNA interference in mammals. EMBO J. 2019, 38, e100941c. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Hammell, C.M.; Ambros, V. Interacting endogenous and exogenous RNAi pathways in Caenorhabditis elegans. RNA 2006, 12, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.J.; Hunter, C.P. The Influence of Competition Among, C. elegans Small RNA Pathways on Development. Genes 2012, 3, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Sarkies, P.; Ashe, A.; Le Pen, J.; McKie, M.A.; Miska, E.A. Competition between virus-derived and endogenous small RNAs regulates gene expression in Caenorhabditis elegans. Genome Res. 2013, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Levanova, A.; Poranen, M.M. RNA Interference as a Prospective Tool for the Control of Human Viral Infections. Front. Microbiol. 2018, 9, 2151. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Singh, K.; Dardick, C.; Kumar Kundu, J. RNAi-Mediated Resistance Against Viruses in Perennial Fruit Plants. Plants 2019, 8, 10. [Google Scholar] [CrossRef]
- Olsvik, P.A.; Williams, T.D.; Tung, H.S.; Mirbahai, L.; Sanden, M.; Skjaerven, K.H.; Ellingsen, S. Impacts of TCDD and MeHg on DNA methylation in zebrafish (Danio rerio) across two generations. Comp. Biochem. Physiol. C Toxicol. Pharm. 2014, 165, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Rudgalvyte, M.; Peltonen, J.; Lakso, M.; Wong, G. Chronic MeHg exposure modifies the histone H3K4me3 epigenetic landscape in Caenorhabditis elegans. Comp. Biochem. Physiol. C Toxicol. Pharm. 2017, 191, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, N.; Cruz, F.; Egea, R.R.; Simon, C.; Sadler-Riggleman, I.; Beck, D.; Nilsson, E.; Ben Maamar, M.; Skinner, M.K. Sperm DNA methylation epimutation biomarker for paternal offspring autism susceptibility. Clin. Epigenet. 2021, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D. Mechanisms for the inheritance of chromatin states. Cell 2011, 146, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, C.B.; Ooi, S.K.; Bestor, T.H.; Bourc’his, D. Epigenetic decisions in mammalian germ cells. Science 2007, 316, 398–399. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 2018, 10, 17. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.; Zhao, L.; Wu, Q.; Wu, C.; Chang, X.; Zhou, Z. Low-Dose Methylmercury-Induced Genes Regulate Mitochondrial Biogenesis via miR-25 in Immortalized Human Embryonic Neural Progenitor Cells. Int. J. Mol. Sci. 2016, 17, 2058. [Google Scholar] [CrossRef] [Green Version]
- Basu, N.; Head, J.; Nam, D.H.; Pilsner, J.R.; Carvan, M.J.; Chan, H.M.; Goetz, F.W.; Murphy, C.A.; Rouvinen-Watt, K.; Scheuhammer, A.M. Effects of methylmercury on epigenetic markers in three model species: Mink, chicken and yellow perch. Comp. Biochem. Physiol. C Toxicol. Pharm. 2013, 157, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Pilsner, J.R.; Lazarus, A.L.; Nam, D.H.; Letcher, R.J.; Sonne, C.; Dietz, R.; Basu, N. Mercury-associated DNA hypomethylation in polar bear brains via the LUminometric Methylation Assay: A sensitive method to study epigenetics in wildlife. Mol. Ecol. 2010, 19, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Go, S.; Kurita, H.; Hatano, M.; Matsumoto, K.; Nogawa, H.; Fujimura, M.; Inden, M.; Hozumi, I. DNA methyltransferase- and histone deacetylase-mediated epigenetic alterations induced by low-level methylmercury exposure disrupt neuronal development. Arch. Toxicol. 2021, 95, 1227–1239. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, T.; Tinkov, A.A.; Skalny, A.V.; Santamaria, A.; Rocha, J.B.T.; Bowman, A.B.; Chen, W.; Aschner, M. Epigenetics and Methylmercury-Induced Neurotoxicity, Evidence from Experimental Studies. Toxics 2023, 11, 72. https://doi.org/10.3390/toxics11010072
Ke T, Tinkov AA, Skalny AV, Santamaria A, Rocha JBT, Bowman AB, Chen W, Aschner M. Epigenetics and Methylmercury-Induced Neurotoxicity, Evidence from Experimental Studies. Toxics. 2023; 11(1):72. https://doi.org/10.3390/toxics11010072
Chicago/Turabian StyleKe, Tao, Alexey A. Tinkov, Anatoly V. Skalny, Abel Santamaria, Joao B. T. Rocha, Aaron B. Bowman, Wen Chen, and Michael Aschner. 2023. "Epigenetics and Methylmercury-Induced Neurotoxicity, Evidence from Experimental Studies" Toxics 11, no. 1: 72. https://doi.org/10.3390/toxics11010072
APA StyleKe, T., Tinkov, A. A., Skalny, A. V., Santamaria, A., Rocha, J. B. T., Bowman, A. B., Chen, W., & Aschner, M. (2023). Epigenetics and Methylmercury-Induced Neurotoxicity, Evidence from Experimental Studies. Toxics, 11(1), 72. https://doi.org/10.3390/toxics11010072