
Effects of Sodium Arsenite on the Myocardial Differentiation in Mouse Embryonic Bodies





Abstract
: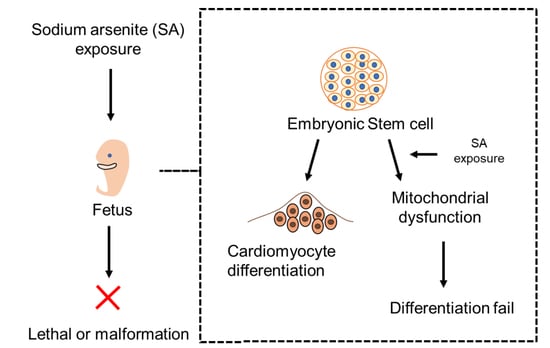
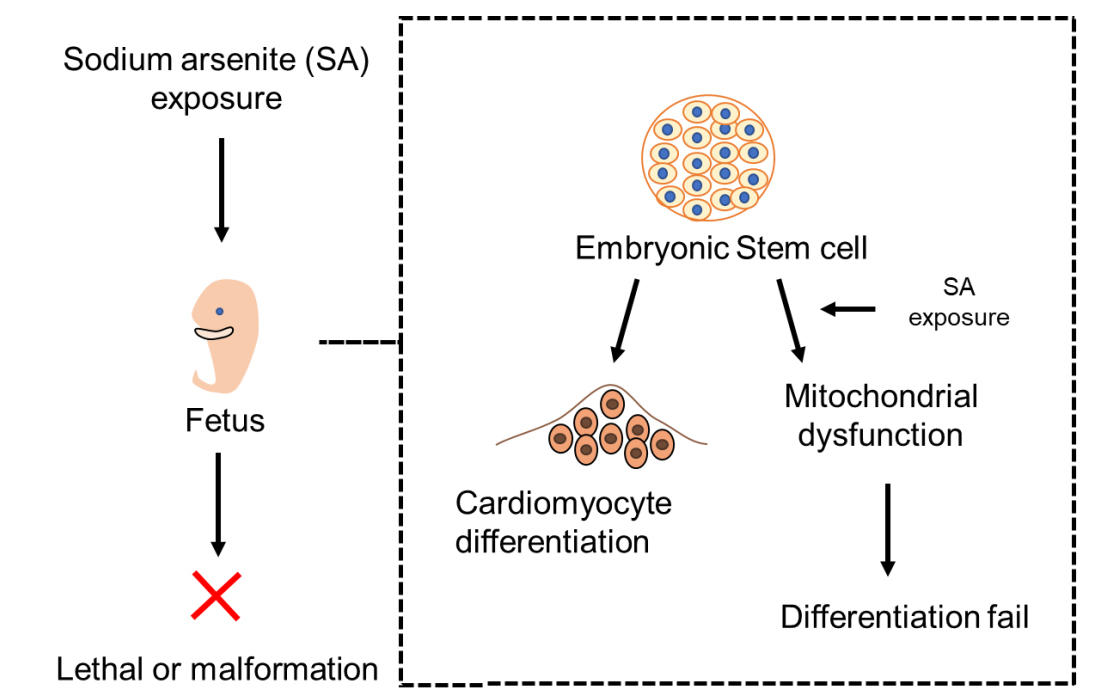{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemical Treatment
2.2. Measurement of Cardiogenic Differentiation in the Embryonic Body
2.3. Analysis of Transcriptional Expression
2.4. Measurement of Mitochondrial Function
2.4.1. Measurement of Mitochondrial ROS (mtROS)
2.4.2. Measurement of Mitochondrial Membrane Potential
2.4.3. Imaging of Live Mitochondria
2.5. Evaluation of Embryotoxicity Using EB Test Method
2.6. Statistics
3. Results
3.1. Sodium Arsenite Disrupted a Myocardial Differentiation of Embryonic Body
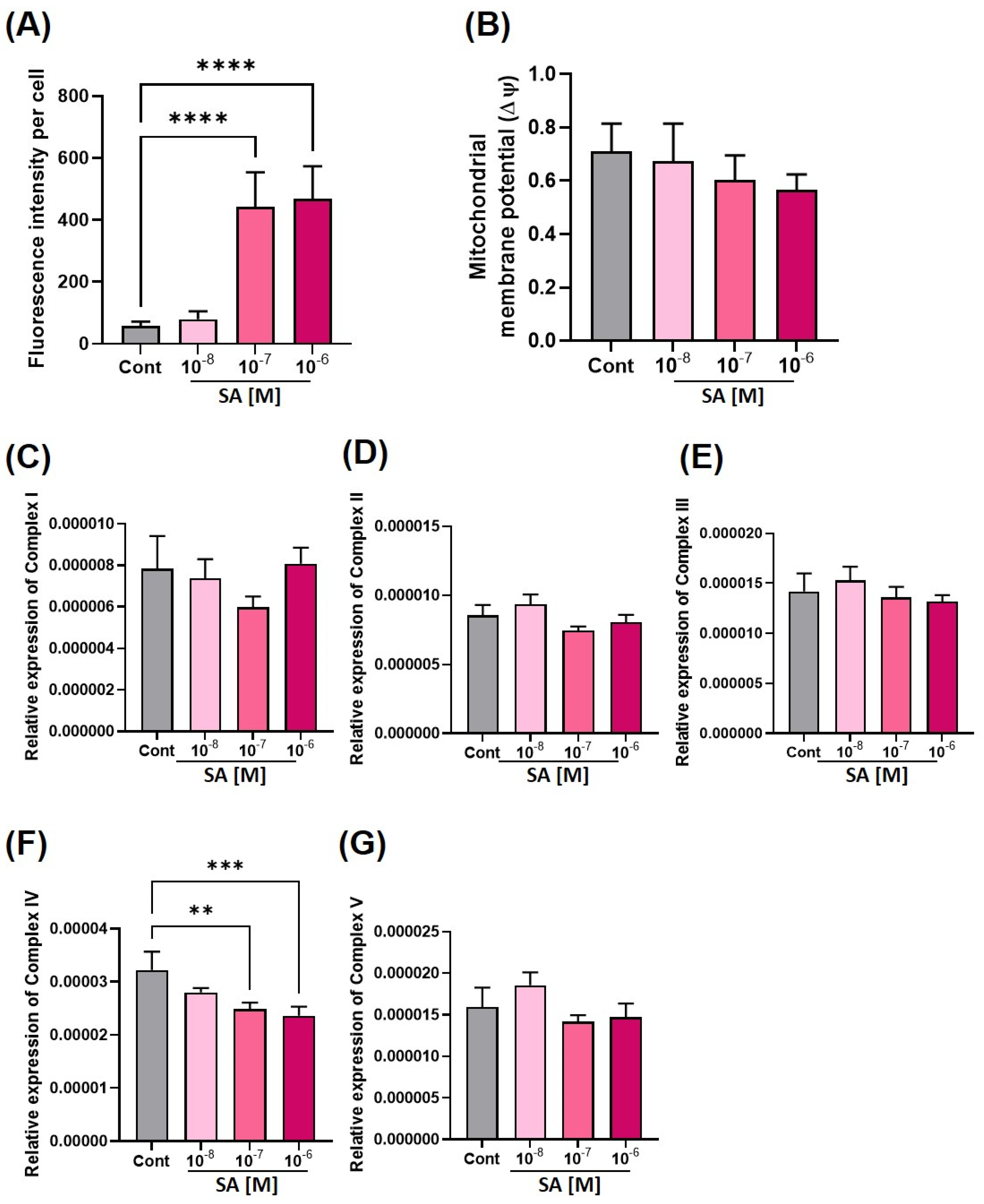
3.2. Effect of Sodium Arsenite on Mitochondria
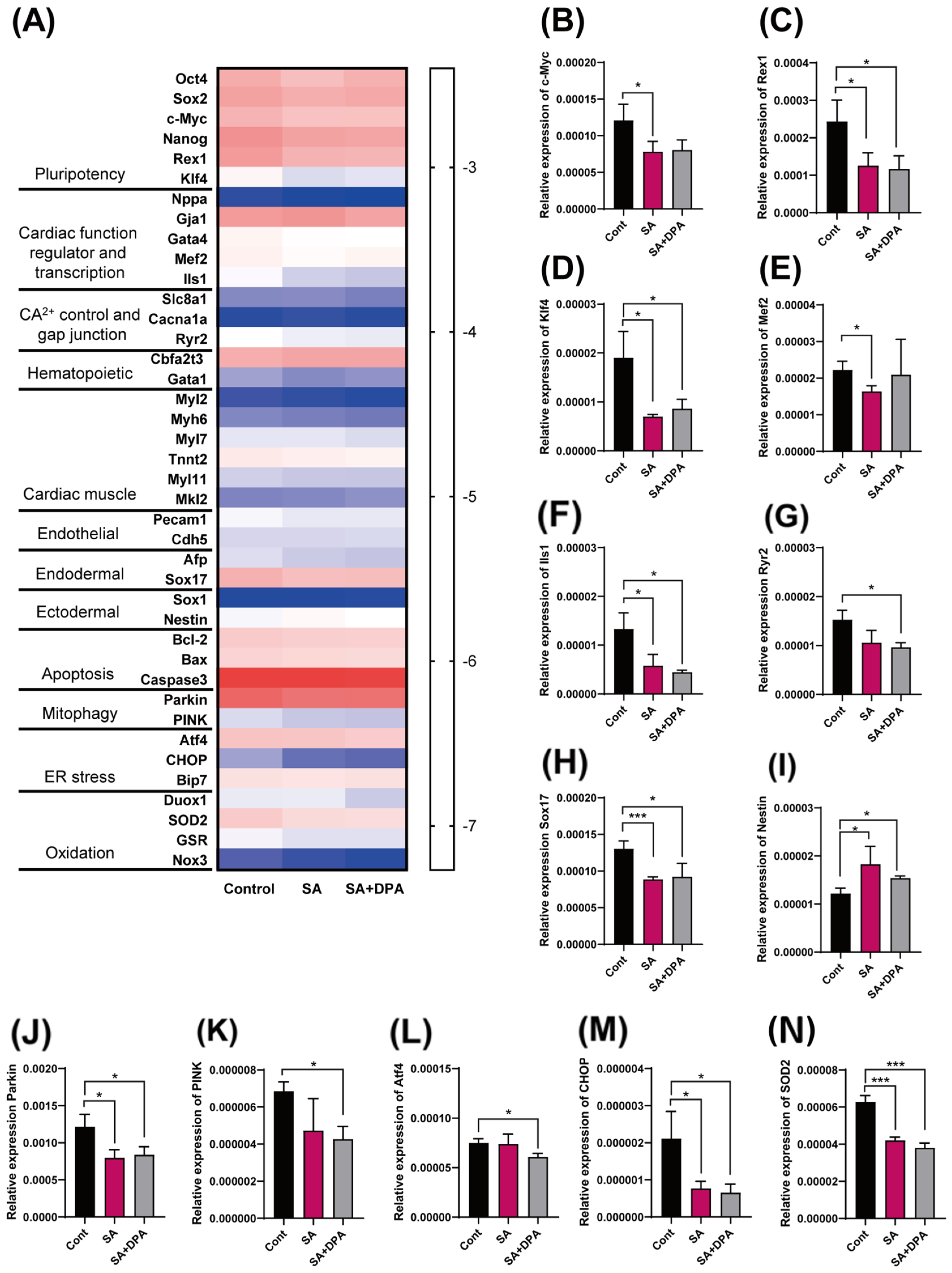
3.3. Effect of D-Penicillamine against Sodium Arsenite during Cardiac Differentiation
3.4. Change in the Gene Transcription Level by Either the SA Treatment or DPA and SA Co-Treatment during the Early EB Development Stage
3.5. Sodium Arsenite Induced Embryotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Upadhyay, M.K.; Shukla, A.; Yadav, P.; Srivastava, S. A review of arsenic in crops, vegetables, animals and food products. Food Chem. 2019, 276, 608–618. [Google Scholar] [CrossRef]
- Winkel, L.H.E.; Pham, T.K.T.; Vi, M.L.; Stengel, C.; Amini, M.; Nguyen, T.H.; Pham, H.V.; Berg, M. Arsenic pollution of groundwater in Vietnam exacerbated by deep aquifer exploitation for more than a century. Proc. Natl. Acad. Sci. USA 2011, 108, 1246–1251. [Google Scholar] [CrossRef]
- Shankar, S.; Shanker, U.; Shikha. Arsenic contamination of groundwater: A review of sources, prevalence, health risks, and strategies for mitigation. Sci. World J. 2014, 2014, 304524. [Google Scholar] [CrossRef]
- Abbas, G.; Murtaza, B.; Bibi, I.; Shahid, M.; Niazi, N.K.; Khan, M.I.; Amjad, M.; Hussain, M.; Natasha. Arsenic Uptake, Toxicity, Detoxification, and Speciation in Plants: Physiological, Biochemical, and Molecular Aspects. Int. J. Environ. Res. Public Health 2018, 15, 59. [Google Scholar] [CrossRef]
- Awasthi, S.; Chauhan, R.; Srivastava, S.; Tripathi, R.D. The Journey of Arsenic from Soil to Grain in Rice. Front. Plant Sci. 2017, 8, 1007. [Google Scholar] [CrossRef]
- Garg, N.; Singla, P. Arsenic toxicity in crop plants: Physiological effects and tolerance mechanisms. Environ. Chem. Lett. 2011, 9, 303–321. [Google Scholar] [CrossRef]
- Ferreccio, C.; Yuan, Y.; Calle, J.; Benítez, H.; Parra, R.L.; Acevedo, J.; Smith, A.H.; Liaw, J.; Steinmaus, C. Arsenic, tobacco smoke, and occupation: Associations of multiple agents with lung and bladder cancer. Epidemiology 2013, 24, 898–905. [Google Scholar] [CrossRef]
- World Health Organization. Guideline for Drinking-Water Quality, 4th ed.; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Ko, I.; Lee, C.H.; Lee, K.P.; Lee, S.W.; Kim, K.W. Remediation of soil contaminated with arsenic, zinc, and nickel by pilot-scale soil washing. Environ. Prog. 2006, 25, 39–48. [Google Scholar] [CrossRef]
- Tsuji, J.S.; Garry, M.R.; Perez, V.; Chang, E.T. Low-level arsenic exposure and developmental neurotoxicity in children: A systematic review and risk assessment. Toxicology 2015, 337, 91–107. [Google Scholar] [CrossRef]
- Felix, K.; Manna, S.K.; Wise, K.; Barr, J.; Ramesh, G.T. Low levels of arsenite activates nuclear factor-kappaB and activator protein-1 in immortalized mesencephalic cells. J. Biochem. Mol. Toxicol. 2005, 19, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.F.; Zheng, B.L.; Bai, J.; Jiang, L.P.; Zheng, Y.; Qi, B.X.; Geng, C.Y.; Zhong, L.F.; Yang, G.; Chen, M.; et al. Low-level sodium arsenite induces apoptosis through inhibiting TrxR activity in pancreatic beta-cells. Environ. Toxicol. Pharmacol. 2015, 40, 486–491. [Google Scholar] [CrossRef]
- Dreval, K.; Tryndyak, V.; Kindrat, I.; Twaddle, N.C.; Orisakwe, O.E.; Mudalige, T.K.; Beland, F.A.; Doerge, D.R.; Pogribny, I.P. Cellular and Molecular Effects of Prolonged Low-Level Sodium Arsenite Exposure on Human Hepatic HepaRG Cells. Toxicol. Sci. 2018, 162, 676–687. [Google Scholar] [CrossRef]
- Wade, T.J.; Xia, Y.J.; Mumford, J.; Wu, K.G.; Le, X.C.; Sams, E.; Sanders, W.E. Cardiovascular disease and arsenic exposure in Inner Mongolia, China: A case control study. Environ. Health 2015, 14, 35. [Google Scholar] [CrossRef]
- Zhao, Q.C.; Sun, Q.C.; Zhou, L.F.; Liu, K.X.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart Assoc. 2019, 8, e012731. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Jiang, W.; Qin, Y.; Gong, G. Mitochondrial Dynamics in Adult Cardiomyocytes and Heart Diseases. Front. Cell Dev. Biol. 2020, 8, 584800. [Google Scholar] [CrossRef]
- Shin, J.W.; Park, S.H.; Kang, Y.G.; Wu, Y.; Choi, H.J.; Shin, J.W. Changes, and the Relevance Thereof, in Mitochondrial Morphology during Differentiation into Endothelial Cells. PLoS ONE 2016, 11, e0161015. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Scime, A. Mitochondrial Function in Muscle Stem Cell Fates. Front. Cell Dev. Biol. 2020, 8, 480. [Google Scholar] [CrossRef]
- Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef]
- Kim, J.I.; Lee, S.Y.; Parka, M.; Kim, S.Y.; Kim, J.W.; Kim, S.A.; Kim, B.N. Peripheral Mitochondrial DNA Copy Number is Increased in Korean Attention-Deficit Hyperactivity Disorder Patients. Front. Psychiatry 2019, 10, 506. [Google Scholar] [CrossRef]
- Rong, Z.Y.; Tu, P.P.; Xu, P.Q.; Sun, Y.; Yu, F.F.; Tu, N.; Guo, L.X.; Yang, Y.N. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Chavan, H.; Christudoss, P.; Mickey, K.; Tessman, R.; Ni, H.M.; Swerdlow, R.; Krishnamurthy, P. Arsenite Effects on Mitochondrial Bioenergetics in Human and Mouse Primary Hepatocytes Follow a Nonlinear Dose Response. Oxidative Med. Cell. Longev. 2017, 12, 9251303. [Google Scholar] [CrossRef]
- Wu, J.; Ni, Y.P.; Wang, X.J.; Yang, Q.L.; Mao, J.Y.; Tong, J.; Zhang, J.; An, Y. Role of mitochondrial DNA in oxidative damage induced by sodium arsenite in human bronchial epithelial cells. J. Toxicol. Environ. Health Part A 2019, 82, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Yih, L.H.; Huang, H.; Jan, K.Y.; Lee, T.C. Sodium Arsenite Induces Atp Depletion and Mitochondrial Damage in Hela-Cells. Cell Biol. Int. Rep. 1991, 15, 253–264. [Google Scholar] [CrossRef]
- Rebuzzini, P.; Cebral, E.; Fassina, L.; Redi, C.A.; Zuccotti, M.; Garagna, S. Arsenic trioxide alters the differentiation of mouse embryonic stem cell into cardiomyocytes. Sci. Rep. 2015, 5, 14993. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, Y.; Lin, Q.X.; Liu, Z.Q.; Wang, H.B.; Duan, C.M.; Wang, Y.M.; Hao, T.; Wu, K.W.; Wang, C.Y. Embryoid bodies formation and differentiation from mouse embryonic stem cells in collagen/Matrigel scaffolds. J. Genet. Genom. 2010, 37, 451–460. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Rohwedel, J.; Hescheler, J.; Wobus, A.M. Embryonic Stem-Cells Differentiate in-Vitro into Cardiomyocytes Representing Sinusnodal, Atrial and Ventricular Cell-Types. Mech. Dev. 1993, 44, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.Y.; Ahn, C.; Yoo, Y.M.; Kim, C.W.; Kim, J.E.; Jo, N.R.; Kang, H.Y.; Jung, E.M.; Kim, K.S.; et al. Second-Phase Validation Study of an Alternative Developmental Toxicity Test Using Mouse Embryonic Stem Cell-Derived Embryoid Bodies. J. Physiol. Pharmacol. 2020, 71, 223–233. [Google Scholar] [CrossRef]
- Kang, H.Y.; Choi, Y.K.; Jo, N.R.; Lee, J.H.; Ahn, C.; Ahn, I.Y.; Kim, T.S.; Kim, K.S.; Choi, K.C.; Lee, J.K.; et al. Advanced developmental toxicity test method based on embryoid body’s area. Reprod. Toxicol. 2017, 72, 74–85. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoo, Y.M.; Lee, B.; Jeong, S.; Tran, D.N.; Jeung, E.B. Melatonin mitigates the adverse effect of hypoxia during myocardial differentiation in mouse embryonic stem cells. J. Vet. Sci. 2021, 22, e54. [Google Scholar] [CrossRef]
- Park, S.M.; Jo, N.R.; Lee, B.; Jung, E.M.; Lee, S.D.; Jeung, E.B. Establishment of a developmental neurotoxicity test by Sox1-GFP mouse embryonic stem cells. Reprod. Toxicol. 2021, 104, 96–105. [Google Scholar] [CrossRef]
- Moore, C.L.; Flanigan, T.J.; Law, C.D.; Loukotkova, L.; Woodling, K.A.; da Costa, G.G.; Fitzpatrick, S.C.; Ferguson, S.A. Developmental neurotoxicity of inorganic arsenic exposure in Sprague-Dawley rats. Neurotoxicol. Teratol. 2019, 72, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia: Possible Interactions with Cellular Processes. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Sun, G.; Cobessi, D.; Wang, A.C.; Shen, J.T.; Tung, E.Y.; Anderson, V.E.; Berry, E.A. 3-nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J. Biol. Chem. 2006, 281, 5965–5972. [Google Scholar] [CrossRef] [PubMed]
- Altas, L.; Isik, M.; Kavurmaci, M. Determination of arsenic levels in the water resources of Aksaray Province, Turkey. J. Environ. Manag. 2011, 92, 2182–2192. [Google Scholar] [CrossRef]
- Ettinger, A.S.; Arbuckle, T.E.; Fisher, M.; Liang, C.L.; Davis, K.; Cirtiu, C.M.; Belanger, P.; LeBlanc, A.; Fraser, W.D.; Grp, M.S. Arsenic levels among pregnant women and newborns in Canada: Results from the Maternal-Infant Research on Environmental Chemicals (MIREC) cohort. Environ. Res. 2017, 153, 8–16. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, X.X.; Yu, H.Y.; Wang, H.; Qi, Y.Y.; Geng, M.Y. Effects of arsenic exposure on d-serine metabolism in the hippocampus of offspring mice at different developmental stages. Arch. Toxicol. 2020, 94, 77–87. [Google Scholar] [CrossRef]
- Kabir, R.; Sinha, P.; Mishra, S.; Ebenebe, O.V.; Taube, N.; Oeing, C.U.; Keceli, G.; Chen, R.; Paolocci, N.; Rule, A.; et al. Inorganic arsenic induces sex-dependent pathological hypertrophy in the heart. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1321–H1336. [Google Scholar] [CrossRef]
- Bao, Z.Y.; Han, Z.B.; Zhang, B.; Yu, Y.; Xu, Z.H.; Ma, W.Y.; Ding, F.Z.; Zhang, L.; Yu, M.X.; Liu, S.Z.; et al. Arsenic trioxide blocked proliferation and cardiomyocyte differentiation of human induced pluripotent stem cells: Implication in cardiac developmental toxicity. Toxicol. Lett. 2019, 309, 51–58. [Google Scholar] [CrossRef]
- Tabacova, S.; Hunter, E.S.; Gladen, B.C. Developmental toxicity of inorganic arsenic in whole embryo culture: Oxidation state, dose, time, and gestational age dependence. Toxicol. Appl. Pharm. 1996, 138, 298–307. [Google Scholar] [CrossRef]
- Hood, R.D.; Vedel, G.C.; Zaworotko, M.J.; Tatum, F.M.; Meeks, R.G. Uptake, Distribution, and Metabolism of Trivalent Arsenic in the Pregnant Mouse. J. Toxicol. Environ. Health 1988, 25, 423–434. [Google Scholar] [CrossRef]
- Rodriguez, V.M.; Carrizales, L.; Mendoza, M.S.; Fajardo, O.R.; Giordano, M. Effects of sodium arsenite exposure on development and behavior in the rat. Neurotoxicol. Teratol. 2002, 24, 743–750. [Google Scholar] [CrossRef]
- Babich, R.; Van Beneden, R.J. Effect of arsenic exposure on early eye development in zebrafish (Danio rerio). J. Appl. Toxicol. 2019, 39, 824–831. [Google Scholar] [CrossRef]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.X. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef]
- Sun, X.C.; Zhan, L.; Chen, Y.B.; Wang, G.; He, L.J.; Wang, Q.; Zhou, F.; Yang, F.; Wu, J.; Wu, Y.S.; et al. Increased mtDNA copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite-stable colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 8. [Google Scholar] [CrossRef]
- Gao, J.; Feng, Z.H.; Wang, X.Q.; Zeng, M.Q.; Liu, J.; Han, S.J.; Xu, J.; Chen, L.; Cao, K.; Long, J.G.; et al. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018, 25, 229–240. [Google Scholar] [CrossRef]
- Evans, N.D.; Minelli, C.; Gentleman, E.; LaPointe, V.; Patankar, S.N.; Kallivretaki, M.; Chen, X.Y.; Roberts, C.J.; Stevens, M.M. Substrate Stiffness Affects Early Differentiation Events in Embryonic Stem Cells. J. Eur. Cells Mater. 2009, 18, 1–13; discussion 13–14. [Google Scholar] [CrossRef] [PubMed]
- Pekkanen-Mattila, M.; Pelto-Huikko, M.; Kujala, V.; Suuronen, R.; Skottman, H.; Aalto-Setala, K.; Kerkela, E. Spatial and temporal expression pattern of germ layer markers during human embryonic stem cell differentiation in embryoid bodies. Histochem. Cell Biol. 2010, 133, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Lolas, M.; Valenzuela, P.D.T.; Tjian, R.; Liu, Z. Charting Brachyury-mediated developmental pathways during early mouse embryogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 4478–4483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, W.A.; Veltri, J.C.; Metcalf, T.J. Acute arsenic exposure treated with oral D-penicillamine. Vet. Hum. Toxicol. 1981, 23, 164–166. [Google Scholar] [PubMed]
- Kumar, N.; Prashant, L.K.; Goyal, V. Wilson’s Disease Update: An Indian Perspective. Ann. Indian Acad. Neurol. 2021, 24, 652–663. [Google Scholar] [CrossRef]
- Mejias, S.G.; Ramphul, K. Penicillamine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Paeezi, M.; Zamani, N.; Hassanian-Moghaddam, H.; Shadnia, S.; Zamani, N.; Chaleshi, V.; Mafi, A.A. Treatment of adult lead poisoning with D-penicillamine. Drug Metab. Pers. Ther. 2019, 34. [Google Scholar] [CrossRef]
- Cao, N.; Liu, Z.; Chen, Z.; Wang, J.; Chen, T.; Zhao, X.; Ma, Y.; Qin, L.; Kang, J.; Wei, B.; et al. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012, 22, 219–236. [Google Scholar] [CrossRef]
- Jung, E.M.; Yoo, Y.M.; Lee, J.H.; Jeung, E.B. Cytotoxicity evaluation and mechanism of endocrine-disrupting chemicals by the embryoid body test. Toxicol. Res. 2022, 38, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Das, J.; Gurunathan, S.; Park, H.W.; Song, H.; Park, C.; Kim, J.H. The cytotoxic effects of dimethyl sulfoxide in mouse preimplantation embryos: A mechanistic study. Theranostics 2017, 7, 4735–4752. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Ahn, C.; Kwon, J.-S.; Kim, K.; Jeung, E.-B. Effects of Sodium Arsenite on the Myocardial Differentiation in Mouse Embryonic Bodies. Toxics 2023, 11, 142. https://doi.org/10.3390/toxics11020142
Jeong S, Ahn C, Kwon J-S, Kim K, Jeung E-B. Effects of Sodium Arsenite on the Myocardial Differentiation in Mouse Embryonic Bodies. Toxics. 2023; 11(2):142. https://doi.org/10.3390/toxics11020142
Chicago/Turabian StyleJeong, SunHwa, Changhwan Ahn, Jin-Sook Kwon, KangMin Kim, and Eui-Bae Jeung. 2023. "Effects of Sodium Arsenite on the Myocardial Differentiation in Mouse Embryonic Bodies" Toxics 11, no. 2: 142. https://doi.org/10.3390/toxics11020142
APA StyleJeong, S., Ahn, C., Kwon, J. -S., Kim, K., & Jeung, E. -B. (2023). Effects of Sodium Arsenite on the Myocardial Differentiation in Mouse Embryonic Bodies. Toxics, 11(2), 142. https://doi.org/10.3390/toxics11020142