Arsenic-Containing Medicine Treatment Disturbed the Human Intestinal Microbial Flora


Abstract
:1. Introduction
2. Material and Methods
2.1. APL Patients
2.2. Sample Collection
2.3. Experimental Flow
2.4. Statistical Analysis of Data
2.5. Bacteroides Fragilis Str. 3783N1-6 Culture and Arsenic Resistance Assays
2.6. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR) Analyses
2.7. Determination of Total Arsenic Concentration
3. Results
3.1. Research Sample Assessment
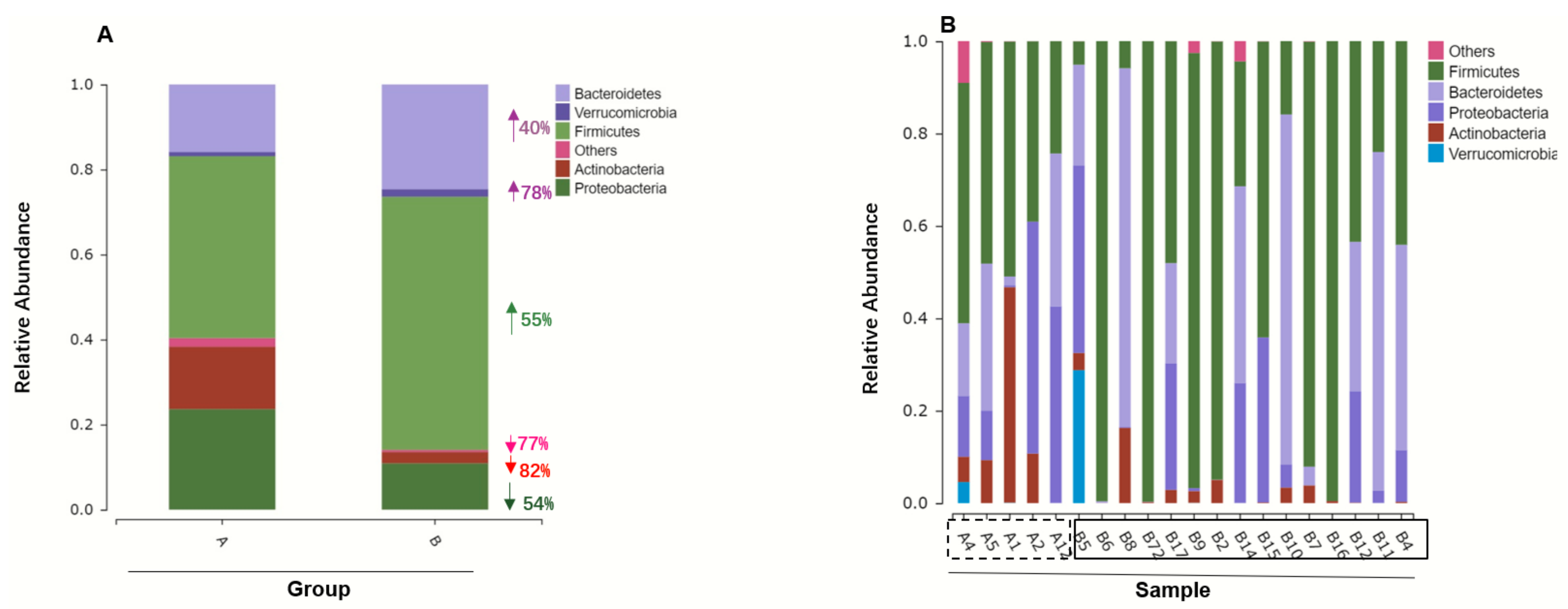
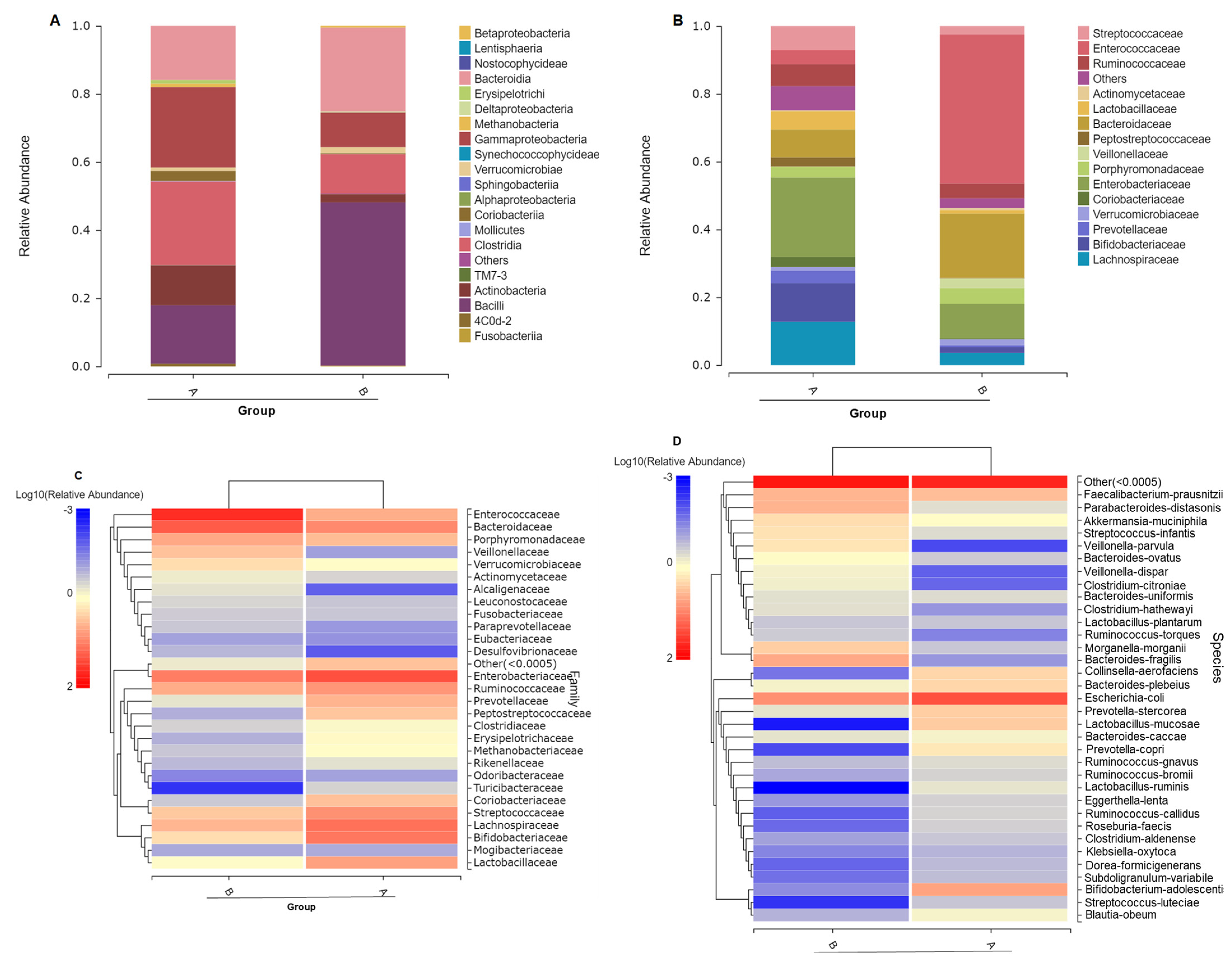
3.2. Gut Microbiome Composition Changes with ATO Treatment
3.3. ATO Treatment Leads to Decrease Microbiome Diversity
3.4. Different Intestinal Microbe Analysis after ATO Treatment Predicts Keystone Taxa Microbe
3.5. Arsenic Resistance Genes Expression in Bacteroides Fragilis
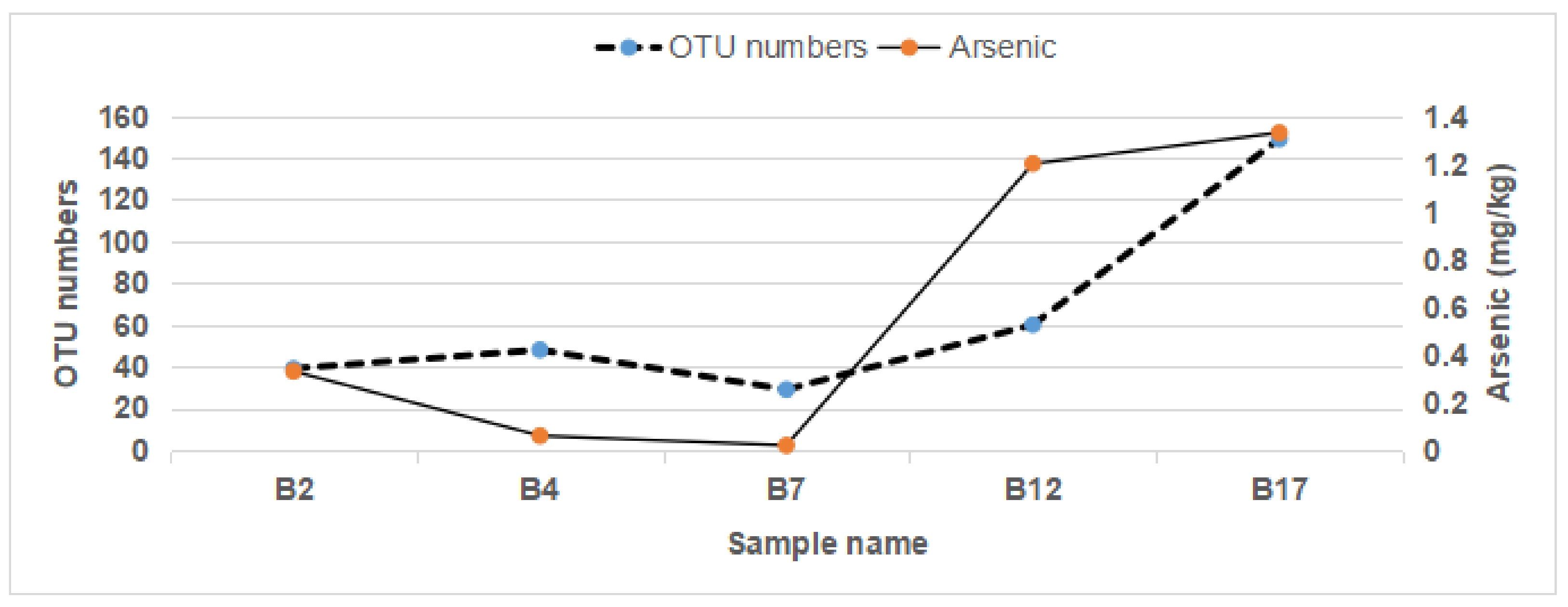
3.6. Gut Microbiome OTU Numbers Were Associated with Arsenic in the Feces
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Y.G.; Yoshinaga, M.; Zhao, F.J.; Rosen, B.P. Earth Abides Arsenic Biotransformations. Annu. Rev. Earth Planet. Sci. 2014, 42, 443–467. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Shen, J.; Rosen, B.P. Pathways of As(III) detoxification in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1999, 96, 5001–5006. [Google Scholar] [CrossRef] [PubMed]
- San Francisco, M.J.; Chen, C.M.; Rosen, B.P. Identification of the membrane component of the anion pump encoded by the arsenical resistance operon of R-factor R773. Prog. Clin. Biol. Res. 1988, 252, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, R.; Bobrowicz, P.; Ułaszewski, S. The Saccharomyces cerevisiae ACR3 Gene Encodes a Putative Membrane Protein Involved in Arsenite Transport. J. Biol. Chem. 1997, 272, 30061–30066. [Google Scholar] [CrossRef]
- Chen, S.-C.; Sun, G.-X.; Yan, Y.; Konstantinidis, K.T.; Zhang, S.-Y.; Deng, Y.; Li, X.-M.; Cui, H.-L.; Musat, F.; Popp, D.; et al. The Great Oxidation Event expanded the genetic repertoire of arsenic metabolism and cycling. Proc. Natl. Acad. Sci. USA 2020, 117, 10414–10421. [Google Scholar] [CrossRef]
- Chen, C.-M.; Misra, T.K.; Silver, S.; Rosen, B.P. Nucleotide Sequence of the Structural Genes for an Anion Pump. J. Biol. Chem. 1986, 261, 15030–15038. [Google Scholar] [CrossRef]
- Liu, Z.; Rensing, C.; Rosen, B.P. Resistance pathways for metalloids and toxic metals. In Metals in Cells; Culotta, V., Scott, R.A., Eds.; Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 429–442. [Google Scholar]
- Lin, Y.-F.; Yang, J.; Rosen, B.P. ArsD: An As(III) metallochaperone for the ArsAB As(III)-translocating ATPase. J. Bioenerg. Biomembr. 2007, 39, 453–458. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Rosen, B.P. Arsenate reductases in prokaryotes and eukaryotes. Environ. Health Perspect. 2002, 110, 745–748. [Google Scholar] [CrossRef]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in medicine: Past, present and future. Biometals 2022, 36, 283–301. [Google Scholar] [CrossRef]
- Thomas, X.; Troncy, J. Arsenic: A beneficial therapeutic poison—A historical overview. Adler Mus. Bull. 2009, 35, 3–13. [Google Scholar]
- Kitareewan, S.; Roebuck, B.D.; Demidenko, E.; Sloboda, R.D.; Dmitrovsky, E. Lysosomes and Trivalent Arsenic Treatment in Acute Promyelocytic Leukemia. Gynecol. Oncol. 2007, 99, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Jurcic, J.G.; Soignet, S.L.; Maslak, P. Diagnosis and treatment of acute promyelocytic leukemia. Curr. Oncol. Rep. 2007, 9, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, B.L.T.; Riemsma, R.; Grimm, S.; Fayter, D.; Deshpande, S.; Armstrong, N.; Witlox, W.; Pouwels, X.; Duffy, S.; Worthy, G.; et al. Arsenic Trioxide for Treating Acute Promyelocytic Leukaemia: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. Pharmacoeconomics 2018, 37, 887–894. [Google Scholar] [CrossRef]
- Antman, K.H. Introduction: The History of Arsenic Trioxide in Cancer Therapy. Oncologgist 2001, 6, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef]
- Cui, X.; Kobayashi, Y.; Hayakawa, T.; Hirano, S. Arsenic Speciation in Bile and Urine Following Oral and Intravenous Exposure to Inorganic and Organic Arsenics in Rats. Toxicol. Sci. 2004, 82, 478–487. [Google Scholar] [CrossRef]
- Shetty, S.A.; Marathe, N.P.; Shouche, Y.S. Opportunities and challenges for gut microbiome studies in the Indian population. Microbiome 2013, 1, 24. [Google Scholar] [CrossRef]
- Lu, K.; Mahbub, R.; Fox, J.G. Xenobiotics: Interaction with the Intestinal Microflora. ILAR J. 2015, 56, 218–227. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Tajkarimi, M.; Wexler, H.M. CRISPR-Cas Systems in Bacteroides fragilis, an Important Pathobiont in the Human Gut Microbiome. Front. Microbiol. 2017, 8, 2234. [Google Scholar] [CrossRef]
- Tamura, K.; Hemsworth, G.R.; DeJean, G.; Rogers, T.E.; Pudlo, N.A.; Urs, K.; Jain, N.; Davies, G.J.; Martens, E.C.; Brumer, H. Molecular Mechanism by which Prominent Human Gut Bacteroidetes Utilize Mixed-Linkage Beta-Glucans, Major Health-Promoting Cereal Polysaccharides. Cell Rep. 2017, 21, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Putnam, E.E.; Abellon-Ruiz, J.; Killinger, B.J.; Rosnow, J.J.; Wexler, A.G.; Folta-Stogniew, E.; Wright, A.T.; van den Berg, B.; Goodman, A.L. Gut Commensal Bacteroidetes Encode a Novel Class of Vitamin B-12-Binding Proteins. mBio 2022, 13, e0284521. [Google Scholar] [CrossRef]
- Lobo, L.A.; Jenkins, A.L.; Smith, C.J.; Rocha, E.R. Expression of Bacteroides fragilis hemolysins in vivo and role of HlyBA in an intra-abdominal infection model. MicrobiologyOpen 2013, 2, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.K.; Mehta, P. Identifying Keystone Species in the Human Gut Microbiome from Metagenomic Timeseries Using Sparse Linear Regression. PLoS ONE 2014, 9, e102451. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Trosvik, P.; de Muinck, E.J. Ecology of bacteria in the human gastrointestinal tract—Identification of keystone and foundation taxa. Microbiome 2015, 3, 44. [Google Scholar] [CrossRef]
- Curtis, M.M.; Hu, Z.; Klimko, C.; Narayanan, S.; Deberardinis, R.; Sperandio, V. The Gut Commensal Bacteroides thetaiotaomicron Exacerbates Enteric Infection through Modification of the Metabolic Landscape. Cell Host Microbe 2014, 16, 759–769. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Deng, H.; Zhong, X.; Li, C.; Luo, Y.; Chen, L.; Zhang, B.; Wang, D.; Huang, Y.; et al. In situ analysis of variations of arsenicals, microbiome and transcriptome profiles along murine intestinal tract. J. Hazard. Mater. 2021, 427, 127899. [Google Scholar] [CrossRef]
- Coryell, M.; McAlpine, M.; Pinkham, N.V.; McDermott, T.R.; Walk, S.T. The gut microbiome is required for full protection against acute arsenic toxicity in mouse models. Nat. Commun. 2018, 9, 5424. [Google Scholar] [CrossRef]
- Van de Wiele, T.; Gallawa, C.M.; Kubachk, K.M.; Creed, J.T.; Basta, N.; Dayton, E.A.; Whitacre, S.; Du Laing, G.; Bradham, K. Arsenic Metabolism by Human Gut Microbiota upon In Vitro Digestion of Contaminated Soils. Environ. Health Perspect. 2010, 118, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Dheer, R.; Patterson, J.; Dudash, M.; Stachler, E.N.; Bibby, K.J.; Stolz, D.B.; Shiva, S.; Wang, Z.; Hazen, S.L.; Barchowsky, A.; et al. Arsenic induces structural and compositional colonic microbiome change and promotes host nitrogen and amino acid metabolism. Toxicol. Appl. Pharmacol. 2015, 289, 397–408. [Google Scholar] [CrossRef]
- Brabec, J.L.; Wright, J.; Ly, T.; Wong, H.T.; McClimans, C.J.; Tokarev, V.; Lamendella, R.; Sherchand, S.; Shrestha, D.; Uprety, S.; et al. Arsenic disturbs the gut microbiome of individuals in a disadvantaged community in Nepal. Heliyon 2020, 6, e03313. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Abo, R.P.; Schlieper, K.A.; Graffam, M.E.; Levine, S.; Wishnok, J.S.; Swenberg, J.A.; Tannenbaum, S.R.; Fox, J.G. Arsenic Exposure Perturbs the Gut Microbiome and Its Metabolic Profile in Mice: An Integrated Metagenomics and Metabolomics Analysis. Environ. Health Perspect. 2014, 122, 284–291. [Google Scholar] [CrossRef]
- Chen, L.; Li, C.; Zhong, X.; Lai, C.; Zhang, B.; Luo, Y.; Guo, H.; Liang, K.; Fang, J.; Zhu, X.; et al. The gut microbiome promotes arsenic metabolism and alleviates the metabolic disorder for their mammal host under arsenic exposure. Environ. Int. 2023, 171, 107660. [Google Scholar] [CrossRef]
- Yu, H.Y.; Wu, B.; Zhang, X.X.; Liu, S.; Yu, J.; Cheng, S.P.; Ren, H.Q.; Ye, L. Arsenic Metabolism and Toxicity Influenced by Ferric Iron in Simulated Gastrointestinal Tract and the Roles of Gut Microbiota. Environ. Sci. Technol. 2016, 50, 7189–7197. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Liang, Z.Z.; Ding, J.; Xue, X.M.; Li, G.; Fu, S.L.; Zhu, D. Arsenic bioaccumulation in the soil fauna alters its gut microbiome and microbial arsenic biotransformation capacity. J. Hazard. Mater. 2021, 417. [Google Scholar] [CrossRef]
- Wang, H.T.; Zhu, D.; Li, G.; Zheng, F.; Ding, J.; O’Connor, P.J.; Zhu, Y.G.; Xue, X.M. Effects of Arsenic on Gut Microbiota and Its Biotransformation Genes in Earthworm Metaphire sieboldi. Environ. Sci. Technol. 2019, 53, 3841–3849. [Google Scholar] [CrossRef]
- Griggs, J.L.; Chi, L.; Hanley, N.M.; Kohan, M.; Herbin-Davis, K.; Thomas, D.J.; Lu, K.; Fry, R.C.; Bradham, K.D. Bioaccessibility of arsenic from contaminated soils and alteration of the gut microbiome in an in vitro gastrointestinal model. Environ. Pollut. 2022, 309, 119753. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for Classification of Acute Leukemias. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Diverio, D.; Rossi, V.; Avvisati, G.; De Santis, S.; Pistilli, A.; Pane, F.; Saglio, G.; Martinelli, G.; Petti, M.C.; Santoro, A.; et al. Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RAR alpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter “AIDA” trial. Blood 1998, 92, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.M.; Macintyre, E.A.; Gabert, J.A.; Delabesse, E.; Rossi, V.; Saglio, G.; Gottardi, E.; Rambaldi, A.; Dotti, G.; Griesinger, F.; et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease—Report of the BIOMED-1 Concerted Action: Investigation of minimal residual disease in acute leukemia. Leukemia 1999, 13, 1901–1928. [Google Scholar] [CrossRef]
- Grimwade, D.; Biondi, A.; Mozziconacci, M.J.; Hagemeijer, A.; Berger, R.; Neat, M.; Howe, K.; Dastugue, N.; Jansen, J.; Radford-Weiss, I.; et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): Results of the European Working Party. Blood 2000, 96, 1297–1308. [Google Scholar] [PubMed]
- Falini, B.; Flenghi, L.; Fagioli, M.; Coco, F.L.; Cordone, I.; Diverio, D.; Pasqualucci, L.; Biondi, A.; Riganelli, D.; Orleth, A.; et al. Immunocytochemical Diagnosis of Acute Promyelocytic Leukemia (M3) With the Monoclonal Antibody PG-M3 (Anti-PML). Blood 1997, 90, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Kumbhare, S.V.; Mhatre, S.S.; Chowdhury, S.P.; Shetty, S.A.; Marathe, N.P.; Bhute, S.; Shouche, Y.S. Exploration of Microbial Diversity and Community Structure of Lonar Lake: The Only Hypersaline Meteorite Crater Lake within Basalt Rock. Front. Microbiol. 2016, 6, 1553. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef]
- Asnicar, F.; Weingart, G.; Tickle, T.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- Xiao, C.; Ran, S.; Huang, Z.; Liang, J. Bacterial Diversity and Community Structure of Supragingival Plaques in Adults with Dental Health or Caries Revealed by 16S Pyrosequencing. Front. Microbiol. 2016, 7, 1145. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Li, J.; Mandal, G.; Rosen, B.P. Expression of arsenic resistance genes in the obligate anaerobe Bacteroides vulgatus ATCC 8482, a gut microbiome bacterium. Anaerobe 2016, 39, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kastl, A.J.; Terry, N.A.; Wu, G.D.; Albenberg, L.G. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2019, 9, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Van de Wouw, M.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. Microbiota-Gut-Brain Axis: Modulator of Host Metabolism and Appetite. J. Nutr. 2017, 147, 727–745. [Google Scholar] [CrossRef]
- Zhou, G.-B.; Li, G.; Chen, S.-J.; Chen, Z. From dissection of disease pathogenesis to elucidation of mechanisms of targeted therapies: Leukemia research in the genomic era. Acta Pharmacol. Sin. 2007, 28, 1434–1449. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.; Jain, R.; Thakur, S.S.; Jain, S.K. Gut Microbiome and Health Assessment Due To Arsenic Toxicity: A Review. Int. J. Life Sci. Pharm. Res. 2021, 11, L52–L60. [Google Scholar]
- Li, D.; Yang, Y.; Li, Y.; Li, Z.; Zhu, X.; Zeng, X. Changes induced by chronic exposure to high arsenic concentrations in the intestine and its microenvironment. Toxicology 2021, 456, 152767. [Google Scholar] [CrossRef]
- Coryell, M.; Roggenbeck, B.A.; Walk, S.T. The Human Gut Microbiome’s Influence on Arsenic Toxicity. Curr. Pharmacol. Rep. 2019, 5, 491–504. [Google Scholar] [CrossRef]
- Niu, S.; Zhu, X.; Zhang, J.; Ma, Y.; Lang, X.; Luo, L.; Li, W.; Zhao, Y.; Zhang, Z. Arsenic trioxide modulates the composition and metabolic function of the gut microbiota in a mouse model of rheumatoid arthritis. Int. Immunopharmacol. 2022, 111, 109159. [Google Scholar] [CrossRef] [PubMed]
- Roggenbeck, B.A.; Banerjee, M.; Leslie, E.M. Cellular arsenic transport pathways in mammals. J. Environ. Sci. 2016, 49, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Twaddle, N.C.; Vanlandingham, M.; Fisher, J.W.; Doerge, D.R. Metabolism and disposition of arsenic species from controlled dosing with sodium arsenite in adult female CD-1 mice. III. Toxicokinetic studies following oral and intravenous administration. Food Chem. Toxicol. 2018, 121, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Droz, P.O.; Vahter, M. A physiologically based pharmacokinetic model for arsenic exposure. 1. Development in hamsters and rabbits. Toxicol. Appl. Pharmacol. 1996, 137, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, T. Experimental Studies on Arsenic Absorption Routes in Rats. Environ. Health Perspect. 1977, 19, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Holladay, S.D.; Wolf, D.C.; Ahmed, S.A.; Robertson, J.L. Reproductive and Developmental Toxicity of Arsenic in Rodents: A Review. Int. J. Toxicol. 2006, 25, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Garcia-Manero, G.; Ferrajoli, A.; Faderl, S.; Verstovsek, S.; Jones, D.; Kantarjian, H. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood 2006, 107, 3469–3473. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, G.; Vogelpoel, L.T.C.; van Capel, T.M.M.; Kapsenberg, M.L.; de Jong, E.C. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol. 2015, 8, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Contreras, A.; Goldfarb, K.C.; Godoy-Vitorino, F.; Karaoz, U.; Contreras, M.; Blaser, M.J.; Brodie, E.L.; Dominguez-Bello, M.G. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2010, 5, 574–579. [Google Scholar] [CrossRef]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Röttjers, L.; Faust, K. Can we predict keystones? Nat. Rev. Microbiol. 2019, 17, 193. [Google Scholar] [CrossRef]
- Makrgeorgou, A.; Leonardi-Bee, J.; Bath-Hextall, F.J.; Murrell, D.F.; Tang, M.L.; Roberts, A.; Boyle, R.J. Probiotics for treating eczema. Cochrane Database Syst. Rev. 2018, 11, Cd006135. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.A.B.; Kim, S.H.; Lee, J.-Y.; Valeriano, V.D.V.; Kang, D.-K. Quantitative Proteogenomics and the Reconstruction of the Metabolic Pathway in Lactobacillus mucosae LM1. Korean J. Food Sci. Anim. Resour. 2015, 35, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Valeriano, V.D.V.; Oh, J.K.; Bagon, B.B.; Kim, H.; Kang, D.-K. Comparative genomic analysis of Lactobacillus mucosae LM1 identifies potential niche-specific genes and pathways for gastrointestinal adaptation. Genomics 2019, 111, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.M.; Stolte, E.H.; London, L.E.E.; Wells, J.M.; Long, S.L.; Joyce, S.A.; Gahan, C.G.M.; Fitzgerald, G.F.; Ross, R.P.; Caplice, N.M.; et al. Lactobacillus mucosae DPC 6426 as a bile-modifying and immunomodulatory microbe. BMC Microbiol. 2019, 19, 33. [Google Scholar] [CrossRef]
- Valeriano, V.D.; Parungao-Balolong, M.M.; Kang, D.K. In vitro evaluation of the mucin-adhesion ability and probiotic potential of Lactobacillus mucosae LM1. J. Appl. Microbiol. 2014, 117, 485–497. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A: Before Arsenic Treatment | C: The Same Patient before Arsenic Treatment | ||
|---|---|---|---|
| B: After Arsenic Treatment | D: The Same Patient after Arsenic Treatment | ||
| Sample Name | Group 1 | Group 2 | Treatment Results |
| A1 | A | no second visit | |
| A12 | A | C | CR |
| A2 | A | C | CR |
| A4 | A | C | CR |
| A5 | A | C | CR |
| B10 | B | CR | |
| B11 | B | CR (+7 chromosome) | |
| B12 | B | D | CR |
| B14 | B | CR | |
| B15 | B | CR | |
| B16 | B | CR | |
| B17 | B | no second visit | |
| B2 | B | D | CR |
| B4 | B | D | CR |
| B5 | B | D | CR |
| B6 | B | CR | |
| B7 | B | CR | |
| B72 | B | CR | |
| B8 | B | DEAD (turn to AML) | |
| B9 | B | CR | |
| B1 | DNA con too low | ||
| A3 | |||
| Levels | Keystone/Functional Species | A (%) | B (%) |
|---|---|---|---|
| Phylum | Euryarchaeota (p = 0.015) Bacteroidetes *** | 0.00% 16.52% | 0.94% 26.27% up |
| Class | Erysipelotrichi (p = 0.011) * Methanobacteria (p = 0.015) * Bacteroidia *** | 1.16% 0.94% 16.52% | 0.13% 0.00% 26.27% up |
| Order | Erysipelotrichales (p = 0.011) * Methanobacteriales (p = 0.015) * Bacteroidales *** | 1.16% 0.94% 16.52% | 0.13% 0.00% 26.27% up |
| Family | Lactobacillacea (p = 0.0083) ** Bacteroidaceae *** | 5.4% 8.92% | 0.95% 20.38% up |
| Genus | Lactobacillus (p = 0.0083) ** Bacteroides *** | 3.6% 8.92% | 0.94% 20.38% up |
| Species | Bifidobacterium adolescentis (p = 0.0043) ** Lactobacillus mucosae (p = 0.023) * Bacteroides fragilis (p = 0.007) ** | 4.9% 2.6% 0.06% | 0.04% 0.002% 4.83% up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chen, X.; Zhao, S.; Chen, J. Arsenic-Containing Medicine Treatment Disturbed the Human Intestinal Microbial Flora. Toxics 2023, 11, 458. https://doi.org/10.3390/toxics11050458
Li J, Chen X, Zhao S, Chen J. Arsenic-Containing Medicine Treatment Disturbed the Human Intestinal Microbial Flora. Toxics. 2023; 11(5):458. https://doi.org/10.3390/toxics11050458
Chicago/Turabian StyleLi, Jiaojiao, Xinshuo Chen, Shixiang Zhao, and Jian Chen. 2023. "Arsenic-Containing Medicine Treatment Disturbed the Human Intestinal Microbial Flora" Toxics 11, no. 5: 458. https://doi.org/10.3390/toxics11050458
APA StyleLi, J., Chen, X., Zhao, S., & Chen, J. (2023). Arsenic-Containing Medicine Treatment Disturbed the Human Intestinal Microbial Flora. Toxics, 11(5), 458. https://doi.org/10.3390/toxics11050458