
Sources, Ionic Composition and Acidic Properties of Bulk and Wet Atmospheric Deposition in the Eastern Middle Adriatic Region

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Measuring Site
2.2. Atmospheric Deposition Sampling
2.3. Analysis of Major Ions in Atmospheric Deposition
2.4. pH Measurements
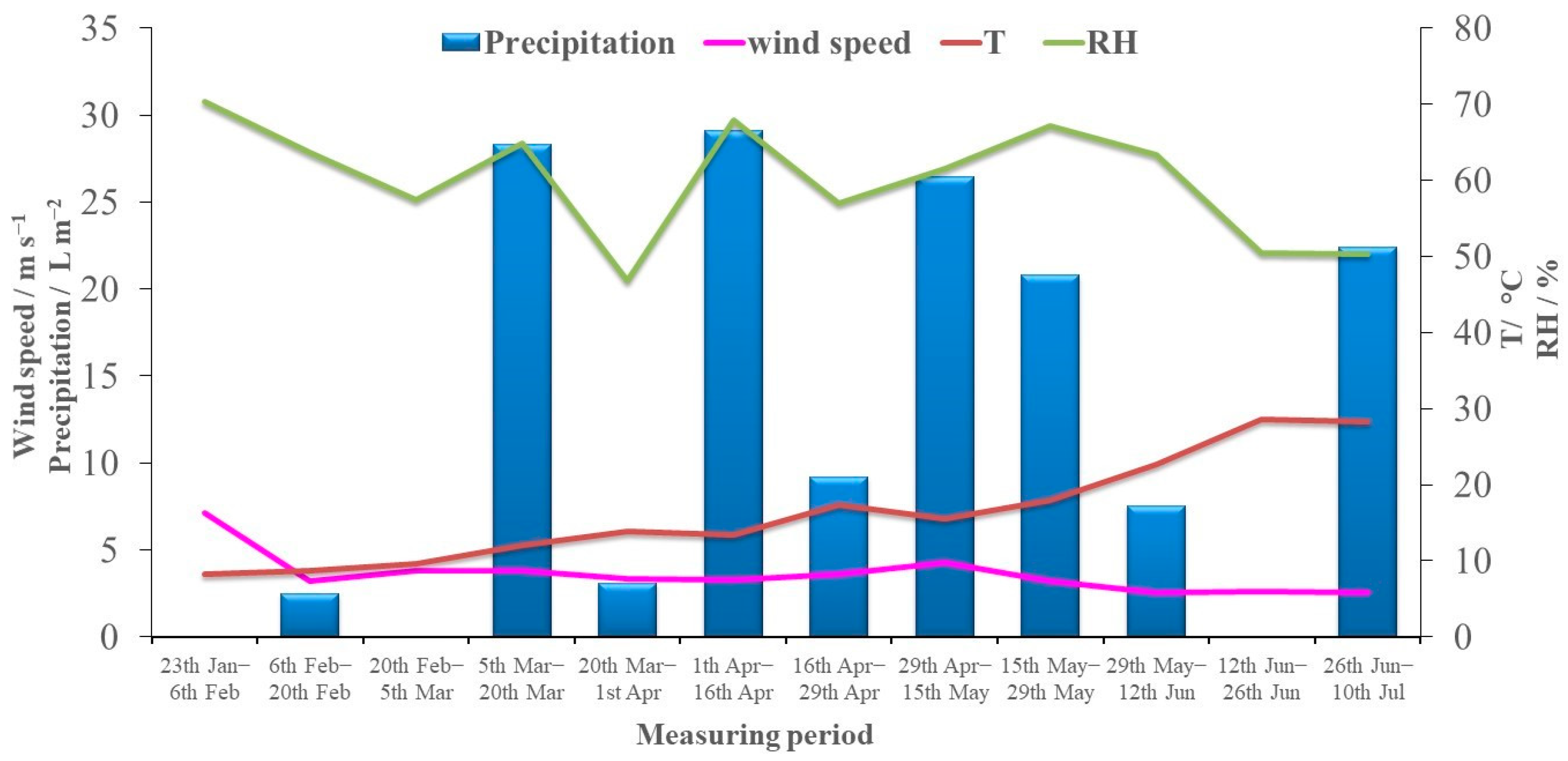
2.5. Meteorological Parameters and Data Analysis
3. Results and Discussion
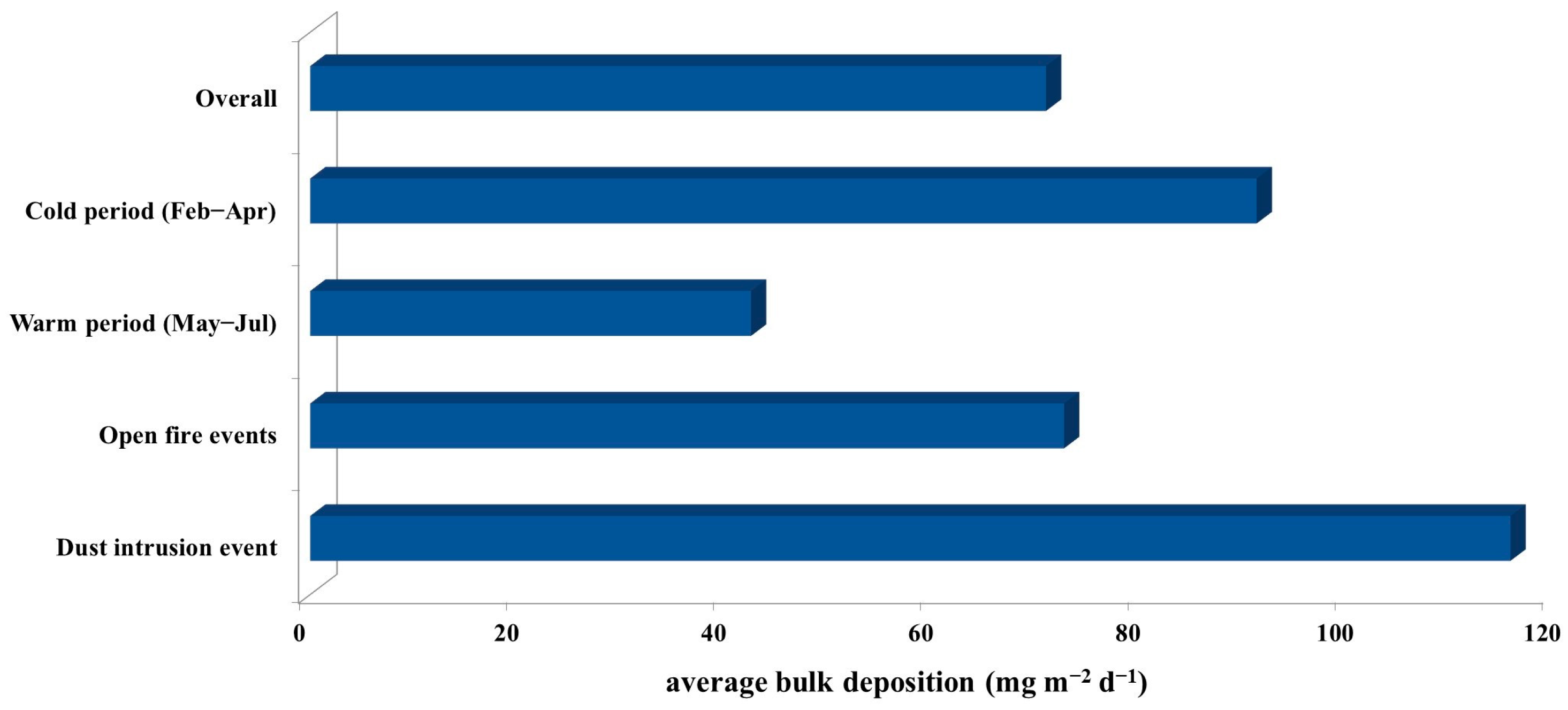
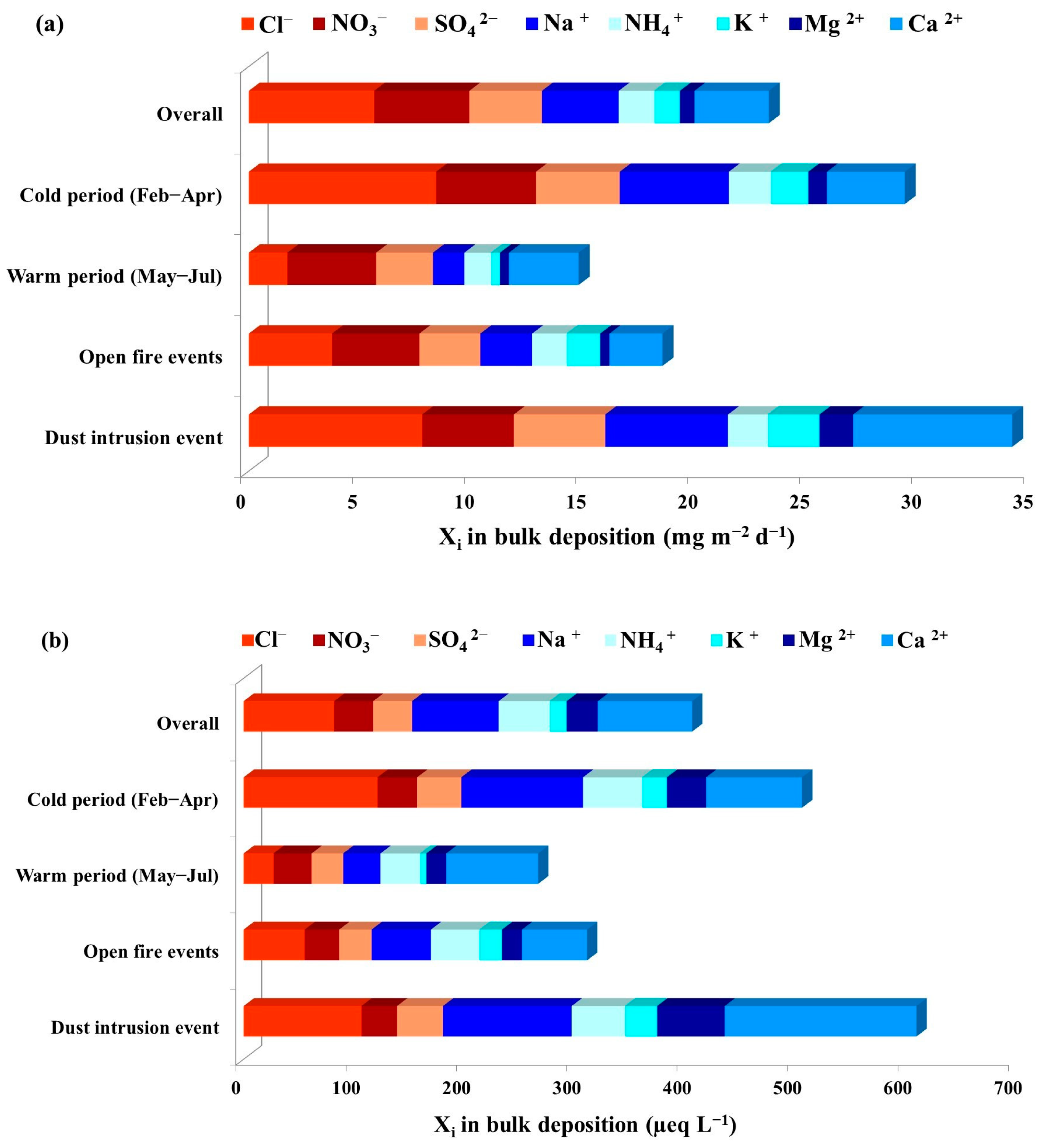
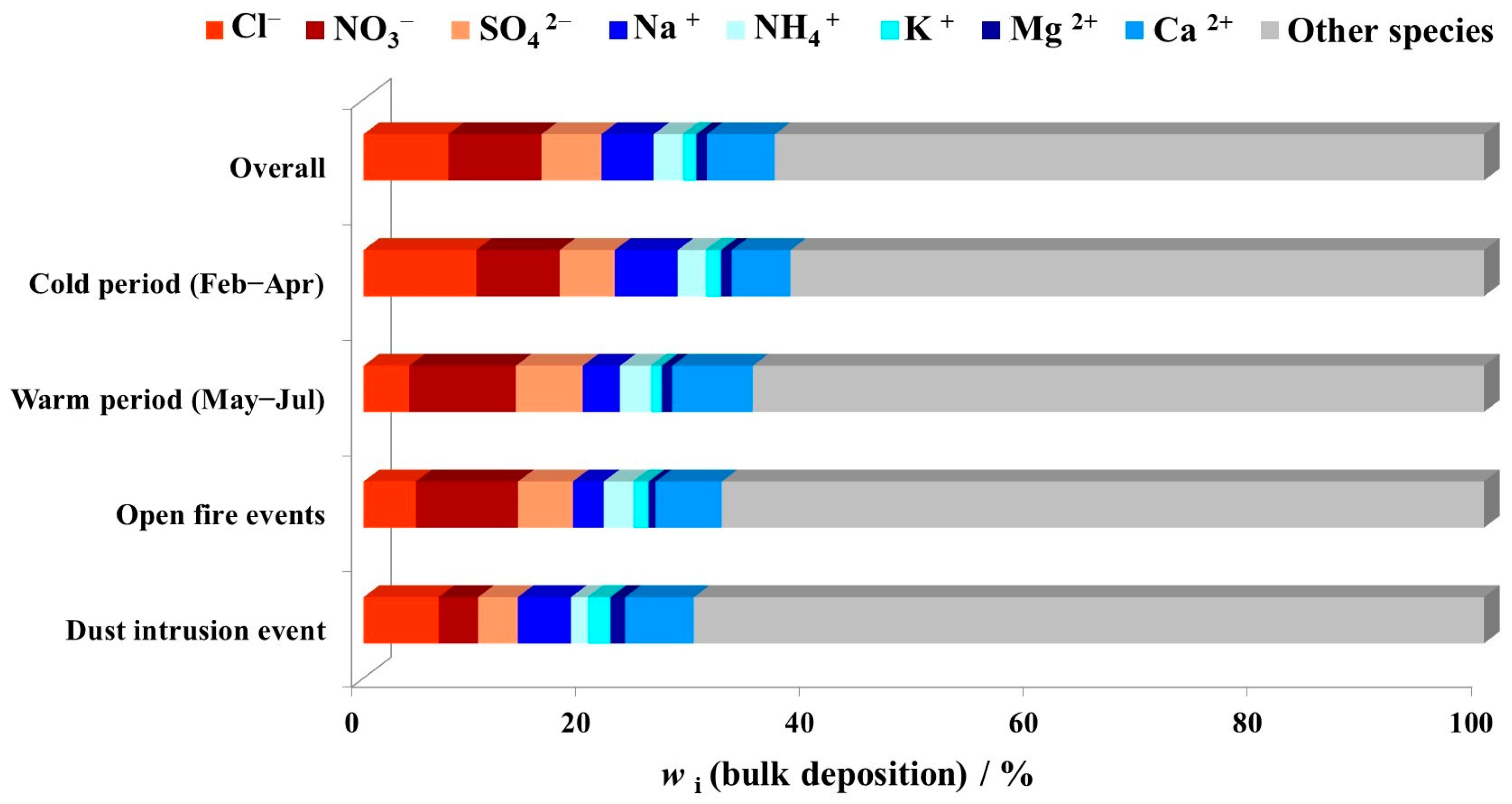
3.1. Major Ions in Bulk Deposition
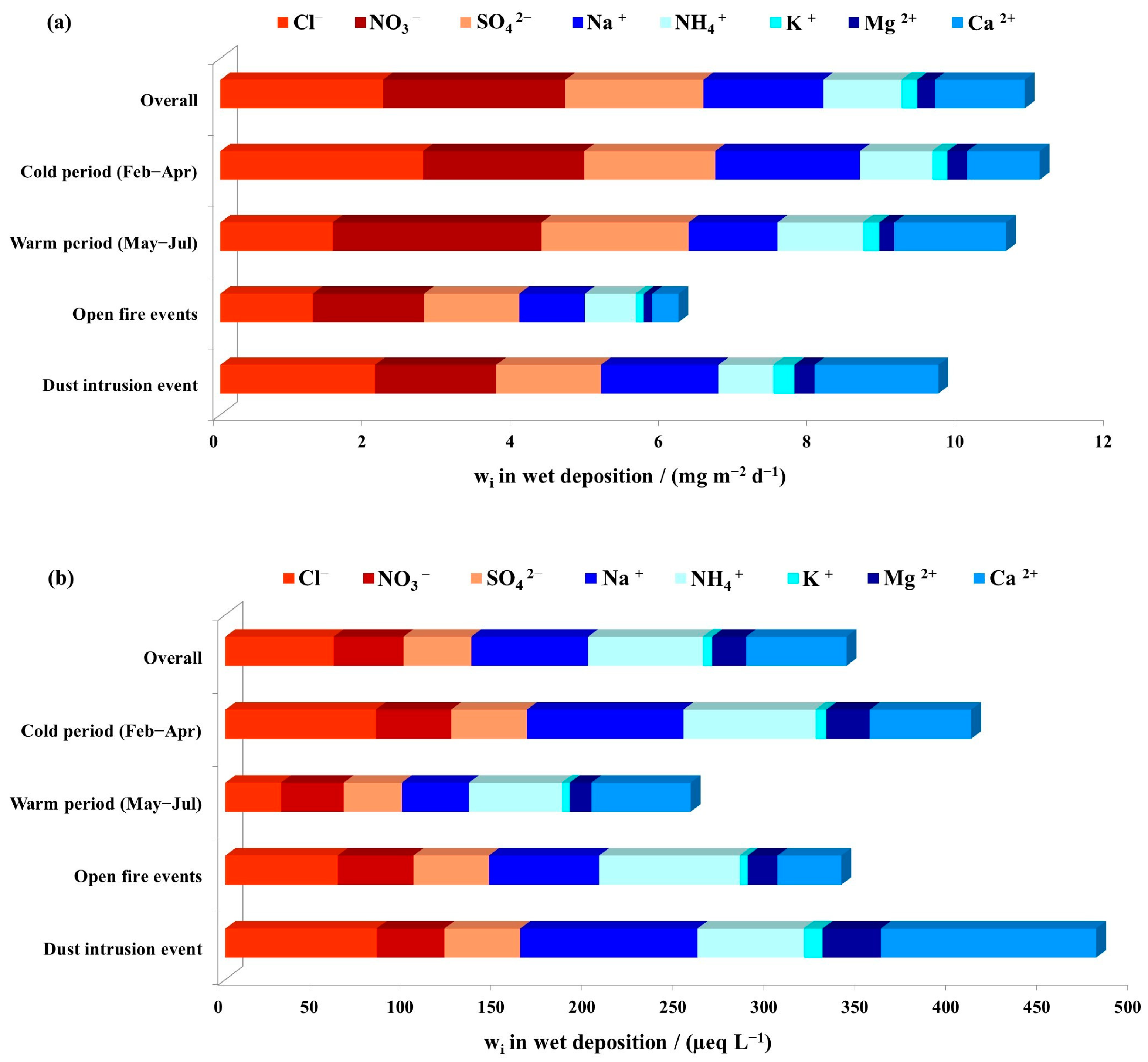
3.2. Major Ions in Wet Deposition
3.3. Acidity and Neutralization Capacity of Atmospheric Deposition
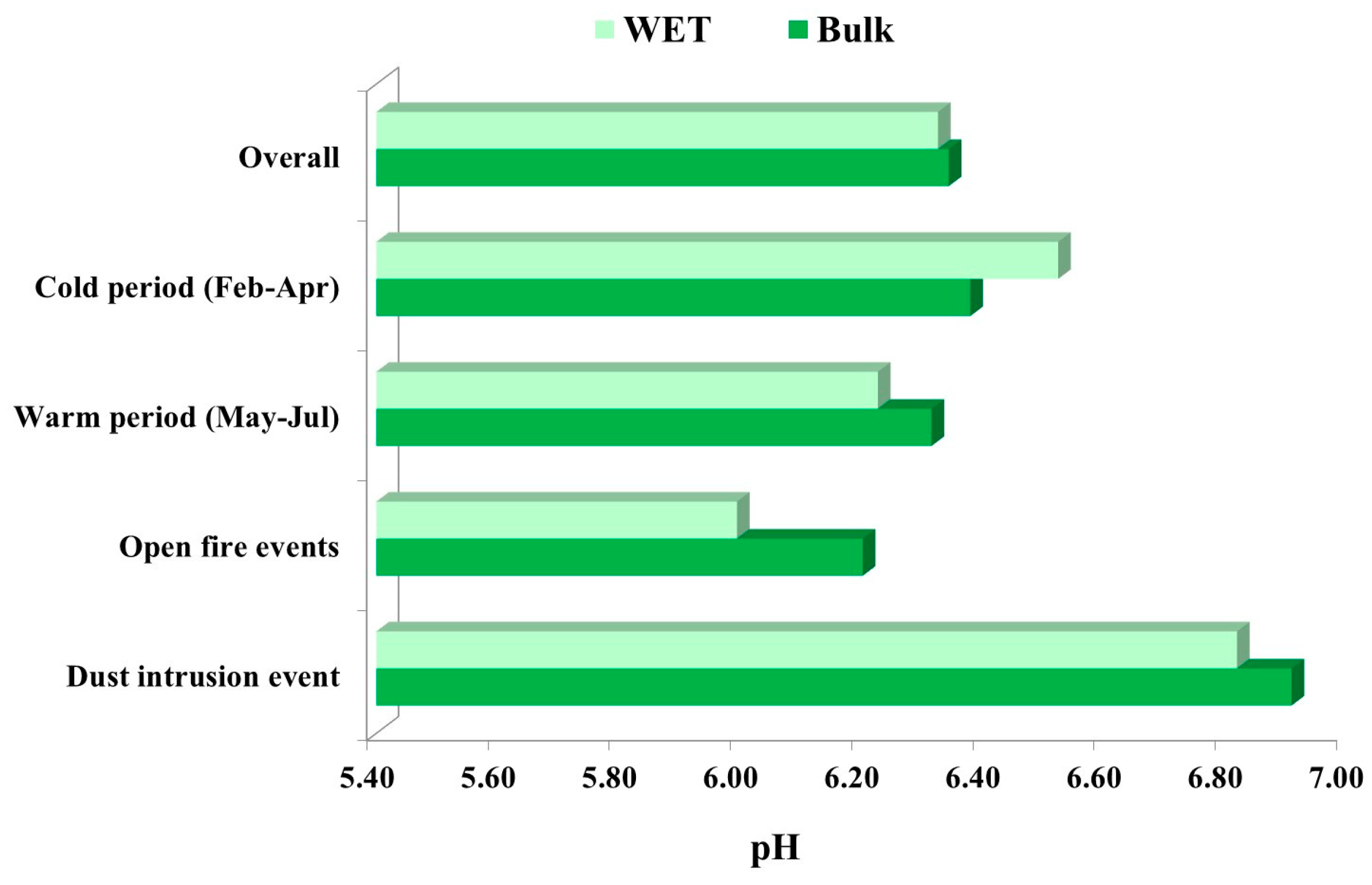
3.3.1. Measurements of Bulk and Wet Deposition pH
3.3.2. Acidity and Neutralization Capacity of Bulk and Wet Deposition
3.4. Dominant Sources of Ions in Atmospheric Deposition
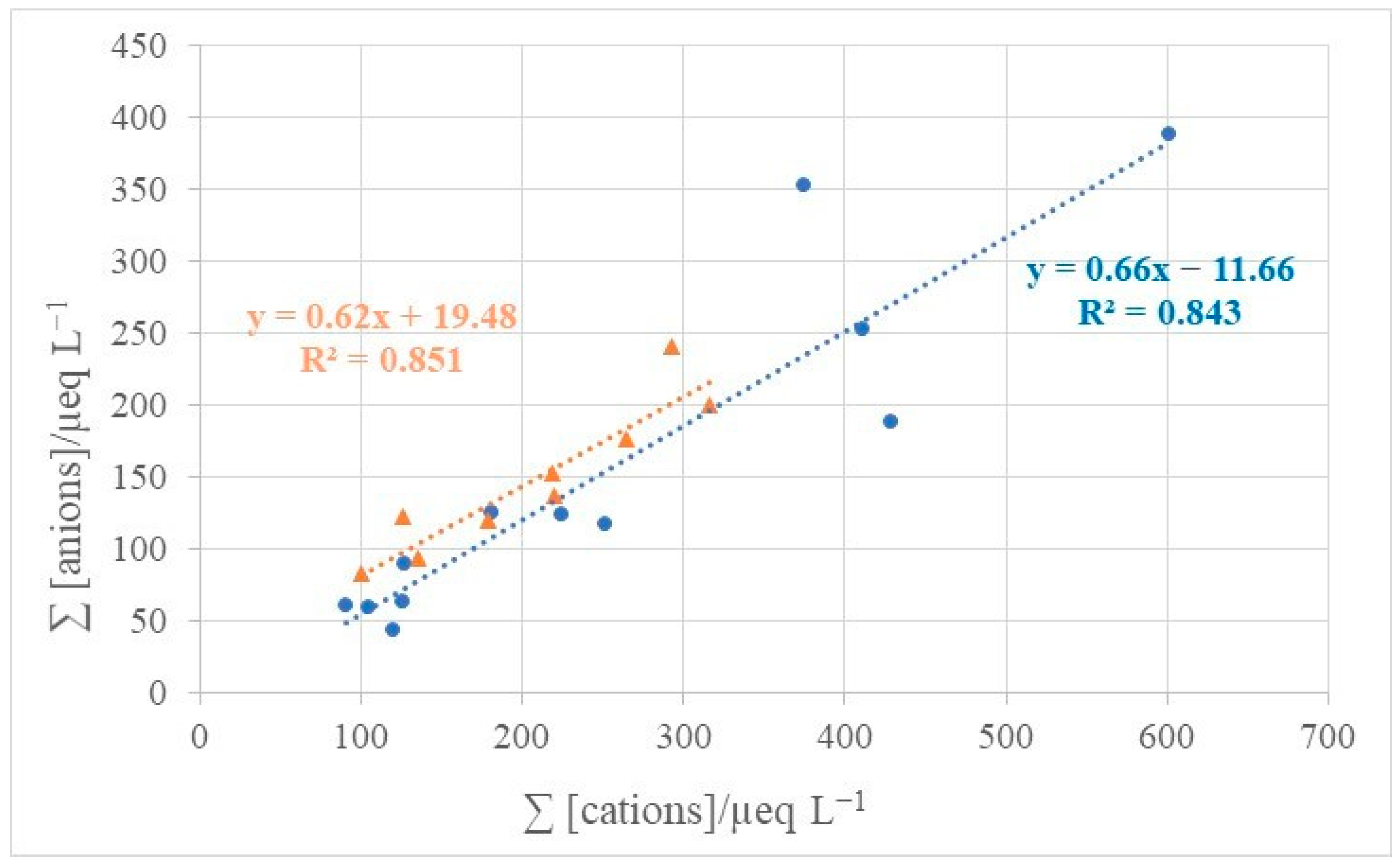
3.4.1. Correlations between Inorganic Compounds in Atmospheric Deposition
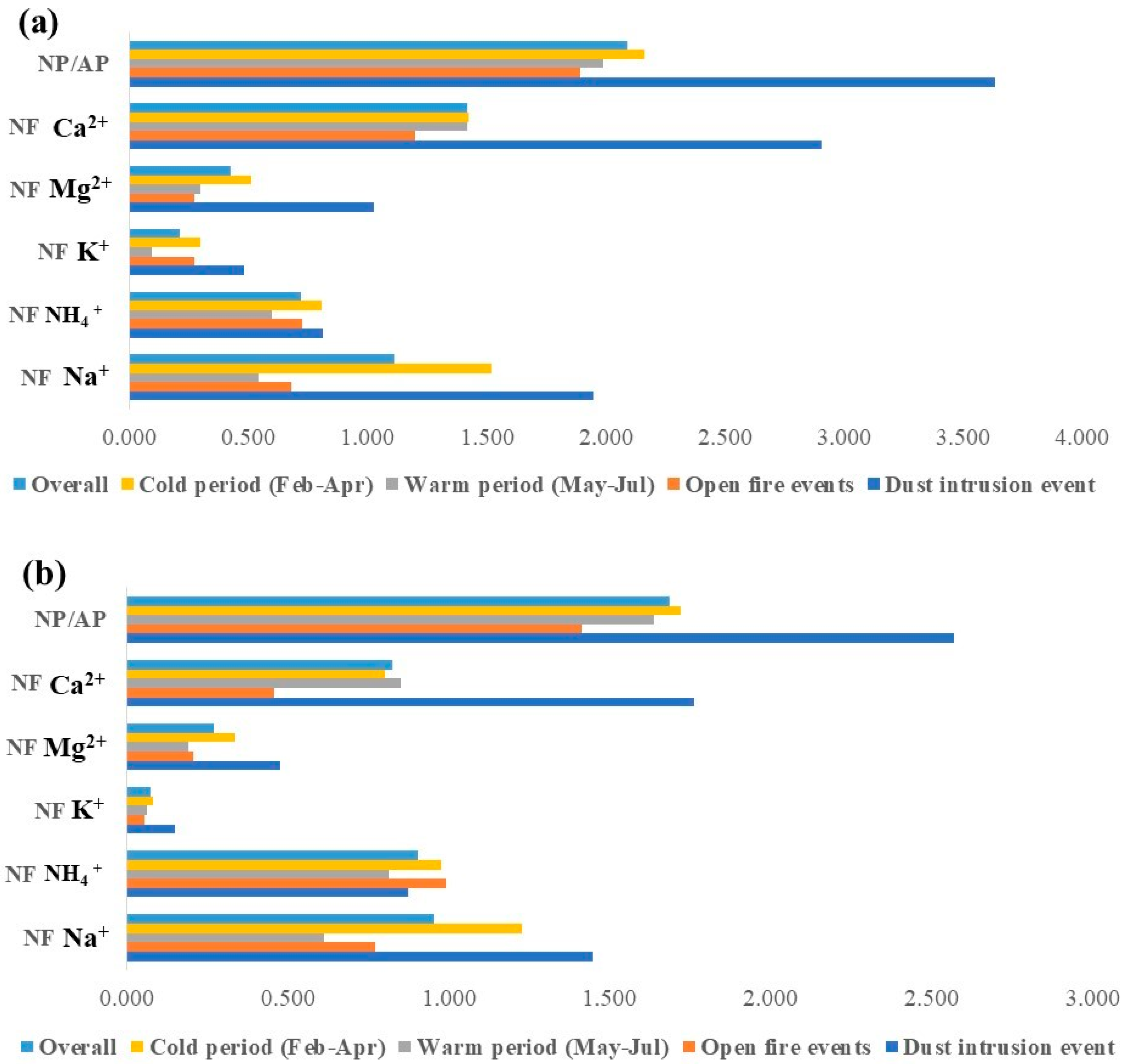
3.4.2. Estimation of Marine and Non-Marine Contribution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morera-Gómez, Y.; Santamaría, J.M.; Elustondo, D.; Lasheras, E.; Alonso-Hernández, C.M. Determination and Source Apportionment of Major and Trace Elements in Atmospheric Bulk Deposition in a Caribbean Rural Area. Atmos. Environ. 2019, 202, 93–104. [Google Scholar] [CrossRef]
- Pereira, J.N.; Fornaro, A.; Vieira-Filho, M. Source Apportionment of Atmospheric Deposition Species in an Agricultural Brazilian Region Using Positive Matrix Factorization. Environ. Sci. Proc. 2021, 8, 12. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Q.; Zhu, J.; Xu, L.; Chen, Z.; Xiao, J.; He, N. Spatiotemporal Variability, Source Apportionment, and Acid-Neutralizing Capacity of Atmospheric Wet Base-Cation Deposition in China. Environ. Pollut. 2020, 262, 114335. [Google Scholar] [CrossRef] [PubMed]
- Querol, X.; Pey, J.; Pandolfi, M.; Alastuey, A.; Cusack, M.; Pérez, N.; Moreno, T.; Viana, M.; Mihalopoulos, N.; Kallos, G.; et al. African Dust Contributions to Mean Ambient PM10 Mass-Levels across the Mediterranean Basin. Atmos. Environ. 2009, 43, 4266–4277. [Google Scholar] [CrossRef]
- Pey, J.; Querol, X.; Alastuey, A.; Forastiere, F.; Stafoggia, M. African Dust Outbreaks over the Mediterranean Basin during 2001–2011: PM10 Concentrations, Phenomenology and Trends, and Its Relation with Synoptic and Mesoscale Meteorology. Atmos. Chem. Phys. 2013, 13, 1395–1410. [Google Scholar] [CrossRef] [Green Version]
- Malaguti, A.; Mircea, M.; La Torretta, T.M.G.; Telloli, C.; Petralia, E.; Stracquadanio, M.; Berico, M. Chemical Composition of Fine and Coarse Aerosol Particles in the Central Mediterranean Area during Dust and Non-Dust Conditions. Aerosol Air Qual. Res. 2015, 15, 410–425. [Google Scholar] [CrossRef] [Green Version]
- Keresztesi, Á.; Birsan, M.V.; Nita, I.A.; Bodor, Z.; Szép, R. Assessing the Neutralisation, Wet Deposition and Source Contributions of the Precipitation Chemistry over Europe during 2000–2017. Environ. Sci. Eur. 2019, 31, 50. [Google Scholar] [CrossRef] [Green Version]
- Keresztesi, Á.; Nita, I.A.; Birsan, M.V.; Bodor, Z.; Pernyeszi, T.; Micheu, M.M.; Szép, R. Assessing the Variations in the Chemical Composition of Rainwater and Air Masses Using the Zonal and Meridional Index. Atmos. Res. 2020, 237, 104846. [Google Scholar] [CrossRef]
- Cerro, J.C.; Cerdà, V.; Caballero, S.; Bujosa, C.; Alastuey, A.; Querol, X.; Pey, J. Chemistry of Dry and Wet Atmospheric Deposition over the Balearic Islands, NW Mediterranean: Source Apportionment and African Dust Areas. Sci. Total Environ. 2020, 747, 141187. [Google Scholar] [CrossRef]
- Pey, J.; Larrasoaña, J.C.; Pérez, N.; Cerro, J.C.; Castillo, S.; Tobar, M.L.; de Vergara, A.; Vázquez, I.; Reyes, J.; Mata, M.P.; et al. Phenomenology and Geographical Gradients of Atmospheric Deposition in Southwestern Europe: Results from a Multi-Site Monitoring Network. Sci. Total Environ. 2020, 744, 140745. [Google Scholar] [CrossRef]
- Izquierdo, R.; Avila, A. Comparison of Collection Methods to Determine Atmospheric Deposition in a Rural Mediterranean Site (NE Spain). J. Atmos. Chem. 2012, 69, 351–368. [Google Scholar] [CrossRef] [Green Version]
- Gambaro, A.; Radaelli, M.; Piazza, R.; Stortini, A.M.; Contini, D.; Belosi, F.; Zangrando, R.; Cescon, P. Organic Micropollutants in Wet and Dry Depositions in the Venice Lagoon. Chemosphere 2009, 76, 1017–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seinfeld John, H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons: Hoboken, NJ, USA, 2016; ISBN 9781119221166. [Google Scholar]
- Niu, Y.; Li, X.; Pu, J.; Huang, Z. Organic Acids Contribute to Rainwater Acidity at a Rural Site in Eastern China. Air Qual. Atmos. Health 2018, 11, 459–469. [Google Scholar] [CrossRef]
- Masood, S.S.; Saied, S.; Siddique, A.; Mohiuddin, S.; Hussain, M.M.; Khan, M.K.; Khwaja, H.A. Influence of Urban–Coastal Activities on Organic Acids and Major Ion Chemistry of Wet Precipitation at a Metropolis in Pakistan. Arab. J. Geosci. 2018, 11, 802. [Google Scholar] [CrossRef]
- Mishra, A.K.; Anshumali. Nature and Sources of Ionic Species in Rainwater during Monsoon Periods in and around Sixteenth–Seventeenth Century CE Monuments in Yamuna River Basin, India. Environ. Monit. Assess. 2021, 193, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, L.; Liu, X.; Zhang, J.; Lin, Y.; Yao, X.; Gao, H.; Zhang, D.; Chen, J.; Wang, W.; et al. Air Pollution–Aerosol Interactions Produce More Bioavailable Iron for Ocean Ecosystems. Sci. Adv. 2017, 3, e1601749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jickells, T.D.; An, Z.S.; Andersen, K.K.; Baker, A.R.; Bergametti, C.; Brooks, N.; Cao, J.J.; Boyd, P.W.; Duce, R.A.; Hunter, K.A.; et al. Global Iron Connections between Desert Dust, Ocean Biogeochemistry, and Climate. Science 2005, 308, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Kanakidou, M.; Duce, R.A.; Prospero, J.M.; Baker, A.R.; Benitez-Nelson, C.; Dentener, F.J.; Hunter, K.A.; Liss, P.S.; Mahowald, N.; Okin, G.S.; et al. Atmospheric Fluxes of Organic N and P to the Global Ocean. Global Biogeochem. Cycles 2012, 26. [Google Scholar] [CrossRef]
- Milinković, A.; Penezić, A.; Kušan, A.C.; Gluščić, V.; Žužul, S.; Skejić, S.; Šantić, D.; Godec, R.; Pehnec, G.; Omanović, D.; et al. Variabilities of Biochemical Properties of the Sea Surface Microlayer: Insights to the Atmospheric Deposition Impacts. Sci. Total Environ. 2022, 838, 156440. [Google Scholar] [CrossRef]
- Paytan, A.; Mackey, K.R.M.; Chen, Y.; Lima, I.D.; Doney, S.C.; Mahowald, N.; Labiosa, R.; Post, A.F. Toxicity of Atmospheric Aerosols on Marine Phytoplankton. Proc. Natl. Acad. Sci. USA 2009, 106, 4601–4605. [Google Scholar] [CrossRef] [Green Version]
- Jordi, A.; Basterretxea, G.; Tovar-Sánchez, A.; Alastuey, A.; Querol, X. Copper Aerosols Inhibit Phytoplankton Growth in the Mediterranean Sea. Proc. Natl. Acad. Sci. USA 2012, 109, 21246–21249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guieu, C.; Loÿe-Pilot, M.D.; Benyahya, L.; Dufour, A. Spatial Variability of Atmospheric Fluxes of Metals (Al, Fe, Cd, Zn and Pb) and Phosphorus over the Whole Mediterranean from a One-Year Monitoring Experiment: Biogeochemical Implications. Mar. Chem. 2010, 120, 164–178. [Google Scholar] [CrossRef]
- Machado, E.C.; Arévalo, P.R.; Casartelli, M.R.O.; Camargo, M.G.; Silva-Filhoc, E.V. Preliminary Study about the Origin of Trace Elements in the Atmospheric Deposition in Two Brazilian Subtropical Estuaries. J. Braz. Chem. Soc. 2016, 27, 735–744. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, H.; Gao, B.; Xu, D. Improved Enrichment Factor Model for Correcting and Predicting the Evaluation of Heavy Metals in Sediments. Sci. Total Environ. 2021, 755, 142437. [Google Scholar] [CrossRef] [PubMed]
- Penezić, A.; Milinković, A.; Bakija Alempijević, S.; Žužul, S.; Frka, S. Atmospheric Deposition of Biologically Relevant Trace Metals in the Eastern Adriatic Coastal Area. Chemosphere 2021, 283, 131178. [Google Scholar] [CrossRef]
- Tsai, Y.I.; Kuo, S.C.; Young, L.H.; Hsieh, L.Y.; Chen, P.T. Atmospheric Dry plus Wet Deposition and Wet-Only Deposition of Dicarboxylic Acids and Inorganic Compounds in a Coastal Suburban Environment. Atmos. Environ. 2014, 89, 696–706. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Peng, L.; Li, Y.; Zhou, Y.; Chai, F.; Mo, Z.; Chen, Z.; Mao, J.; Wang, W. Characterization of Precipitation in the Background of Atmospheric Pollutants Reduction in Guilin: Temporal Variation and Source Apportionment. J. Environ. Sci. 2020, 98, 1–13. [Google Scholar] [CrossRef]
- Shen, Z.; Zhang, L.; Cao, J.; Tian, J.; Liu, L.; Wang, G.; Zhao, Z.; Wang, X.; Zhang, R.; Liu, S. Chemical Composition, Sources, and Deposition Fluxes of Water-Soluble Inorganic Ions Obtained from Precipitation Chemistry Measurements Collected at an Urban Site in Northwest China. J. Environ. Monit. 2012, 14, 3000–3008. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, X.; Zhao, P.; Shi, Z.; Harrison, R.M. PM2.5 Source Apportionment Using Organic Marker-Based Chemical Mass Balance Modeling: Influence of Inorganic Markers and Sensitivity to Source Profiles. Atmos. Environ. 2023, 294, 119477. [Google Scholar] [CrossRef]
- Vet, R.; Artz, R.S.; Carou, S.; Shaw, M.; Ro, C.U.; Aas, W.; Baker, A.; Bowersox, V.C.; Dentener, F.; Galy-Lacaux, C.; et al. A Global Assessment of Precipitation Chemistry and Deposition of Sulfur, Nitrogen, Sea Salt, Base Cations, Organic Acids, Acidity and PH, and Phosphorus. Atmos. Environ. 2014, 93, 3–100. [Google Scholar] [CrossRef]
- Calvo-Fernández, J.; Marcos, E.; Calvo, L. Bulk Deposition of Atmospheric Inorganic Nitrogen in Mountainous Heathland Ecosystems in North-Western Spain. Atmos. Res. 2017, 183, 237–244. [Google Scholar] [CrossRef]
- Calvo, A.I.; Olmo, F.J.; Lyamani, H.; Alados-Arboledas, L.; Castro, A.; Fernández-Raga, M.; Fraile, R. Chemical Composition of Wet Precipitation at the Background EMEP Station in Víznar (Granada, Spain) (2002–2006). Atmos. Res. 2010, 96, 408–420. [Google Scholar] [CrossRef]
- Pieri, L.; Matzneller, P.; Gaspari, N.; Marotti, I.; Dinelli, G.; Rossi, P. Bulk Atmospheric Deposition in the Southern Po Valley (Northern Italy). Water Air. Soil Pollut. 2010, 210, 155–169. [Google Scholar] [CrossRef]
- Morabito, E.; Contini, D.; Belosi, F.; Stortini, A.M.; Manodori, L.; Gambaro, A. Atmospheric Deposition of Inorganic Elements and Organic Compounds at the Inlets of the Venice Lagoon. Adv. Meteorol. 2014, 2014, 158902. [Google Scholar] [CrossRef] [Green Version]
- Yatkin, S.; Adali, M.; Bayram, A. A Study on the Precipitation in Izmir, Turkey: Chemical Composition and Source Apportionment by Receptor Models. J. Atmos. Chem. 2016, 73, 241–259. [Google Scholar] [CrossRef]
- Čačković, M.; Kalinić, N.; Vadjić, V.; Pehnec, G. Heavy Metals and Acidic Components in Total Deposited Matter in Šibenik and National Park Kornati, Croatia. Arch. Environ. Contam. Toxicol. 2009, 56, 12–20. [Google Scholar] [CrossRef]
- Jakovljević, I.; Šimić, I.; Mendaš, G.; Sever Štrukil, Z.; Žužul, S.; Gluščić, V.; Godec, R.; Pehnec, G.; Bešlić, I.; Milinković, A.; et al. Pollution Levels and Deposition Processes of Airborne Organic Pollutants over the Central Adriatic Area: Temporal Variabilities and Source Identification. Mar. Pollut. Bull. 2021, 172, 112873. [Google Scholar] [CrossRef]
- Orlović-Leko, P.; Plavšić, M.; Bura-Nakić, E.; Kozarac, Z.; Ćosović, B. Organic Matter in the Bulk Precipitations in Zagreb and Šibenik, Croatia. Atmos. Environ. 2009, 43, 805–811. [Google Scholar] [CrossRef]
- Periš, N.; Buljac, M.; Bralić, M.; Buzuk, M.; Brinić, S.; Plazibat, I. Characterization of the Air Quality in Split, Croatia Focusing Upon Fine and Coarse Particulate Matter Analysis. Anal. Lett. 2015, 48, 553–565. [Google Scholar] [CrossRef]
- Cvitešić Kušan, A.; Kroflič, A.; Grgić, I.; Ciglenečki, I.; Frka, S. Chemical Characterization of Fine Aerosols in Respect to Water-Soluble Ions at the Eastern Middle Adriatic Coast. Environ. Sci. Pollut. Res. 2020, 27, 10249–10264. [Google Scholar] [CrossRef]
- Frka, S.; Dautović, J.; Kozarac, Z.; Ćosović, B.; Hoffer, A.; Kiss, G. Surface-Active Substances in Atmospheric Aerosol: An Electrochemical Approach. Tellus Ser. B Chem. Phys. Meteorol. 2012, 64, 18490. [Google Scholar] [CrossRef] [Green Version]
- Jakovljević, I.; Sever Štrukil, Z.; Pehnec, G.; Bešlić, I.; Milinković, A.; Bakija Alempijević, S.; Frka Milosavljević, S. Comparison of PAH Mass Concentrations in Aerosols of the Middle Adriatic Coast Area and Central Croatia. Kem. Ind. 2020, 69, 75–82. [Google Scholar] [CrossRef]
- Šiljković, Ž.; Mamut, M. Forest Fires in Dalmatia. Bull. Geogr. Socio-Econ. Ser. 2016, 32, 117–130. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Environmental Health Criteria 54 “Ammonia”; World Health Organization: Geneva, Switzerland, 1986. [Google Scholar]
- Ivančan-Picek, B. (Ed.) DHMZ Meteorološki i Hidrološki Bilten; DHMZ: Zagreb, Croatia, 2019; ISSN 1334-3017. [Google Scholar]
- Narodne Novine, Official Gazette 77/2020, Croatian Regulation on Levels of Pollutants in Ambient Air; Narodne Novine: Zagreb, Croatia, 2020; Volume 77.
- Đorđević, D.S.; Tošić, I.; Unkašević, M.; Đurašković, P. Water-Soluble Main Ions in Precipitation over the Southeastern Adriatic Region: Chemical Composition and Long-Range Transport. Environ. Sci. Pollut. Res. 2010, 17, 1591–1598. [Google Scholar] [CrossRef]
- Kara, M.; Dumanoglu, Y.; Altiok, H.; Elbir, T.; Odabasi, M.; Bayram, A. Seasonal and Spatial Variations of Atmospheric Trace Elemental Deposition in the Aliaga Industrial Region, Turkey. Atmos. Res. 2014, 149, 204–216. [Google Scholar] [CrossRef]
- Rossini, P.; Guerzoni, S.; Molinaroli, E.; Rampazzo, G.; De Lazzari, A.; Zancanaro, A. Atmospheric Bulk Deposition to the Lagoon of Venice: Part I. Fluxes of Metals, Nutrients and Organic Contaminants. Environ. Int. 2005, 31, 959–974. [Google Scholar] [CrossRef] [Green Version]
- Lemou, A.; Rabhi, L.; Merabet, H.; Ladji, R.; Nicolas, J.B.; Bonnaire, N.; Mustapha, M.A.; Dilmi, R.; Sciare, J.; Mihalopoulos, N.; et al. Chemical Characterization of Fine Particles (PM2.5) at a Coastal Site in the South Western Mediterranean during the ChArMex Experiment. Environ. Sci. Pollut. Res. 2020, 27, 20427–20445. [Google Scholar] [CrossRef] [PubMed]
- Safai, P.D.; Rao, P.S.P.; Momin, G.A.; Ali, K.; Chate, D.M.; Praveen, P.S. Chemical Composition of Precipitation during 1984-2002 at Pune, India. Atmos. Environ. 2004, 38, 1705–1714. [Google Scholar] [CrossRef]
- Kumar, R.; Elizabeth, A.; Gawane, A.G. Air Quality Profile of Inorganic Ionic Composition of Fine Aerosols at Two Sites in Mumbai City. Aerosol Sci. Technol. 2006, 40, 477–489. [Google Scholar] [CrossRef]
- Gao, J.; Li, Y.; Li, J.; Shi, G.; Liu, Z.; Han, B.; Tian, X.; Wang, Y.; Feng, Y.; Russell, A.G. Impact of Formation Pathways on Secondary Inorganic Aerosol During Haze Pollution in Beijing: Quantitative Evidence From High-Resolution Observation and Modeling. Geophys. Res. Lett. 2021, 48, e2021GL095623. [Google Scholar] [CrossRef]
- Alebic-Juretic, A.; Mifka, B. Secondary Sulfur and Nitrogen Species in PM10 from the Rijeka Bay Area (Croatia). Bull. Environ. Contam. Toxicol. 2017, 98, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Cerro, J.C.; Cerdà, V.; Querol, X.; Alastuey, A.; Bujosa, C.; Pey, J. Variability of Air Pollutants, and PM Composition and Sources at a Regional Background Site in the Balearic Islands: Review of Western Mediterranean Phenomenology from a 3-Year Study. Sci. Total Environ. 2020, 717, 137177. [Google Scholar] [CrossRef]
- Huy, D.H.; Thanh, L.T.; Hien, T.T.; Takenaka, N. Comparative Study on Water-Soluble Inorganic Ions in PM2.5 from Two Distinct Climate Regions and Air Quality. J. Environ. Sci. 2020, 88, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Hu, B.; Li, Y.; Shi, G. Investigating Atmospheric Nitrate Sources and Formation Pathways between Heating and Non-Heating Seasons in Urban North China. Environ. Res. Lett. 2023, 18, 034006. [Google Scholar] [CrossRef]
- Ge, X.; He, Y.; Sun, Y.; Xu, J.; Wang, J.; Shen, Y.; Chen, M. Characteristics and Formation Mechanisms of Fine Particulate Nitrate in Typical Urban Areas in China. Atmosphere 2017, 8, 62. [Google Scholar] [CrossRef] [Green Version]
- Marić, D.; Frka, S.; Godrijan, J.; Tomažić, I.; Penezić, A.; Djakovac, T.; Vojvodić, V.; Precali, R.; Gašparović, B. Organic Matter Production during Late Summer-Winter Period in a Temperate Sea. Cont. Shelf Res. 2013, 55, 52–65. [Google Scholar] [CrossRef]
- Anatolaki, C.; Tsitouridou, R. Relationship between Acidity and Ionic Composition of Wet Precipitation. A Two Years Study at an Urban Site, Thessaloniki, Greece. Atmos. Res. 2009, 92, 100–113. [Google Scholar] [CrossRef]
- Ma, L.; Dadashazar, H.; Hilario, M.R.A.; Cambaliza, M.O.; Lorenzo, G.R.; Simpas, J.B.; Nguyen, P.; Sorooshian, A. Contrasting Wet Deposition Composition between Three Diverse Islands and Coastal North American Sites. Atmos. Environ. 2021, 244, 117919. [Google Scholar] [CrossRef]
- Chathuranga, R.A.J.; Liyandeniya, A.B.; Dharmapriya, T.N.; Deeyamulla, M.P.; Priyantha, N. Risk Assessment and Source Apportionment of Wet Bulk Deposition in Three Typical Sites of Gampaha District, Sri Lanka. SN Appl. Sci. 2020, 2, 1394. [Google Scholar] [CrossRef]
- Xu, Z.; Wu, Y.; Liu, W.J.; Liang, C.S.; Ji, J.; Zhao, T.; Zhang, X. Chemical Composition of Rainwater and the Acid Neutralizing Effect at Beijing and Chizhou City, China. Atmos. Res. 2015, 164, 278–285. [Google Scholar] [CrossRef]
- Xing, J.; Song, J.; Yuan, H.; Li, X.; Li, N.; Duan, L.; Qu, B.; Wang, Q.; Kang, X. Chemical Characteristics, Deposition Fluxes and Source Apportionment of Precipitation Components in the Jiaozhou Bay, North China. Atmos. Res. 2017, 190, 10–20. [Google Scholar] [CrossRef]
- Sosa Echeverría, R.; Alarcón Jiménez, A.L.; del Carmen Torres Barrera, M.; Sánchez Alvarez, P.; Granados Hernandez, E.; Vega, E.; Jaimes Palomera, M.; Retama, A.; Gay, D.A. Nitrogen and Sulfur Compounds in Ambient Air and in Wet Atmospheric Deposition at Mexico City Metropolitan Area. Atmos. Environ. 2023, 292, 119411. [Google Scholar] [CrossRef]
- Meng, Y.; Zhao, Y.; Li, R.; Li, J.; Cui, L.; Kong, L.; Fu, H. Characterization of Inorganic Ions in Rainwater in the Megacity of Shanghai: Spatiotemporal Variations and Source Apportionment. Atmos. Res. 2019, 222, 12–24. [Google Scholar] [CrossRef]
- Valappil, N.K.M.; Viswanathan, P.M.; Hamza, V. Chemical Characteristics of Rainwater in the Tropical Rainforest Region in Northwestern Borneo. Environ. Sci. Pollut. Res. 2020, 27, 36994–37010. [Google Scholar] [CrossRef]
- Conradie, E.H.; Van Zyl, P.G.; Pienaar, J.J.; Beukes, J.P.; Galy-Lacaux, C.; Venter, A.D.; Mkhatshwa, G.V. The Chemical Composition and Fluxes of Atmospheric Wet Deposition at Four Sites in South Africa. Atmos. Environ. 2016, 146, 113–131. [Google Scholar] [CrossRef] [Green Version]
- Pu, W.; Quan, W.; Ma, Z.; Shi, X.; Zhao, X.; Zhang, L.; Wang, Z.; Wang, W. Long-Term Trend of Chemical Composition of Atmospheric Precipitation at a Regional Background Station in Northern China. Sci. Total Environ. 2017, 580, 1340–1350. [Google Scholar] [CrossRef] [Green Version]
- Brugnone, F.; D’Alessandro, W.; Parello, F.; Liotta, M.; Bellomo, S.; Prano, V.; Li Vigni, L.; Sprovieri, M.; Calabrese, S. Atmospheric Deposition around the Industrial Areas of Milazzo and Priolo Gargallo (Sicily–Italy)—Part A: Major Ions. Int. J. Environ. Res. Public Health 2023, 20, 3898. [Google Scholar] [CrossRef]
- Karydis, V.A.; Tsimpidi, A.P.; Pozzer, A.; Lelieveld, J. How Alkaline Compounds Control Atmospheric Aerosol Particle Acidity. Atmos. Chem. Phys. 2021, 21, 14983–15001. [Google Scholar] [CrossRef]
- Roy, A.; Chatterjee, A.; Tiwari, S.; Sarkar, C.; Das, S.K.; Ghosh, S.K.; Raha, S. Precipitation Chemistry over Urban, Rural and High Altitude Himalayan Stations in Eastern India. Atmos. Res. 2016, 181, 44–53. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Victor, T.; Chun, N. Chemical and Statistical Analysis of Precipitation in Singapore. Water Air. Soil Pollut. 2001, 130, 451–456. [Google Scholar] [CrossRef]
- Pascaud, A.; Sauvage, S.; Coddeville, P.; Nicolas, M.; Croisé, L.; Mezdour, A.; Probst, A. Contrasted Spatial and Long-Term Trends in Precipitation Chemistry and Deposition Fluxes at Rural Stations in France. Atmos. Environ. 2016, 146, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Hsu, L.F.; Huang, S.Z.; Zheng, L. Assessment of the Components and Sources of Acid Deposition in Northeast Asia: A Case Study of the Coastal and Metropolitan Cities in Northern Taiwan. Atmosphere 2020, 11, 983. [Google Scholar] [CrossRef]
- Mansour, K.; Decesari, S.; Bellacicco, M.; Marullo, S.; Santoleri, R.; Bonasoni, P.; Facchini, M.C.; Ovadnevaite, J.; Ceburnis, D.; O’Dowd, C.; et al. Particulate Methanesulfonic Acid over the Central Mediterranean Sea: Source Region Identification and Relationship with Phytoplankton Activity. Atmos. Res. 2020, 237, 104837. [Google Scholar] [CrossRef]
- UNEP; MAP; MEDPOL. Atmospheric Transport and Deposition of Pollutants into the Mediterranean Sea Final Reports on Research Projects; World Meteorological Organization Map Technical Reports Series No. 133; UNEP: Athens, Greece, 2001; ISBN 9280720708. [Google Scholar]
- Gillett, R.W.; Ayers, G.P. Rainwater acidity at Jabiru, Australia, in the wet season of 1983/84. Sci. Total Environ. 1990, 92, 129–144. [Google Scholar] [CrossRef]
- Pehnec, G.; Jakovljević, I.; Godec, R.; Sever Štrukil, Z.; Žero, S.; Huremović, J.; Džepina, K. Carcinogenic Organic Content of Particulate Matter at Urban Locations with Different Pollution Sources. Sci. Total Environ. 2020, 734, 139414. [Google Scholar] [CrossRef]
- Keene, W.C.; Pszenny, A.A.P.; Galloway, J.N.; Hawley, M.E. Sea-Salt Corrections and Interpretation of Constituent Ratios in Marine Precipitation. J. Geophys. Res. 1986, 91, 6647. [Google Scholar] [CrossRef]
- World Health Organization. WHO Global Air Quality Guidelines; World Health Organization: Geneva, Switzerland, 2021; ISBN 978-92-4-003421-1. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
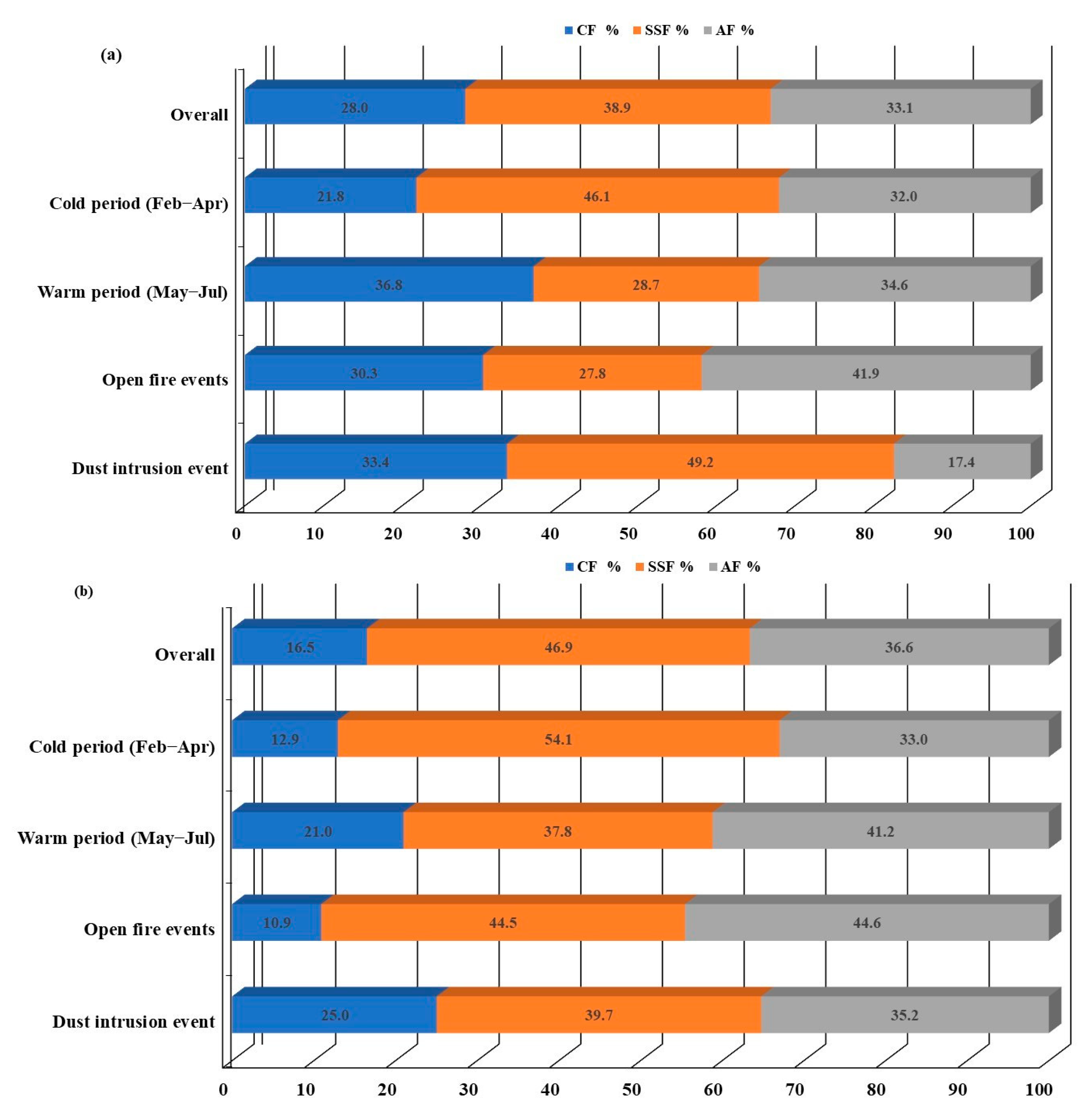{kind=link}
| Deposition Flux/mg m−2 d−1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Statistical Parameter | Bulk Deposition | Cl− | NO3− | SO42− | Na+ | NH4+ | K+ | Mg2+ | Ca2+ |
| Cmin | 22 | 0.15 | 3.04 | 1.39 | 0.20 | 0.65 | 0.11 | 0.12 | 1.70 |
| Cmax | 185 | 19.44 | 5.36 | 7.54 | 10.19 | 4.05 | 5.75 | 1.50 | 7.11 |
| Cavg | 71 | 5.61 | 4.25 | 3.25 | 3.43 | 1.61 | 1.13 | 0.65 | 3.33 |
| SD | 50 | 6.32 | 0.78 | 1.79 | 3.46 | 0.89 | 1.63 | 0.52 | 1.63 |
| concentration/µeq L−1 | |||||||||
| Cmin | 2.25 | 25.81 | 15.21 | 4.66 | 18.96 | 1.31 | 4.57 | 38.29 | |
| Cmax | 265.07 | 48.71 | 88.54 | 249.69 | 126.88 | 83.12 | 64.41 | 173.79 | |
| Cavg | 82.09 | 35.30 | 35.28 | 78.43 | 46.57 | 15.14 | 28.14 | 85.46 | |
| SD | 92.64 | 8.21 | 21.20 | 81.22 | 28.89 | 23.14 | 22.33 | 42.00 | |
| Deposition Flux/mg m−2 d−1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Statistical Parameter | Cl− | NO3− | SO42− | Na+ | NH4+ | K+ | Mg2+ | Ca2+ |
| Cmin | 0.41 | 0.59 | 0.47 | 0.28 | 0.34 | 0.04 | 0.04 | 0.15 |
| Cmax | 7.00 | 4.38 | 3.28 | 4.92 | 1.99 | 0.45 | 0.66 | 2.71 |
| Cavg | 2.19 | 2.46 | 1.86 | 1.62 | 1.06 | 0.21 | 0.24 | 1.21 |
| SD | 2.03 | 1.45 | 1.15 | 1.44 | 0.60 | 0.15 | 0.19 | 0.95 |
| concentration/µeq L−1 | ||||||||
| Cmin | 21.41 | 27.58 | 19.79 | 22.95 | 28.00 | 2.37 | 7.21 | 16.10 |
| Cmax | 129.01 | 53.55 | 58.33 | 113.38 | 104.71 | 10.11 | 32.01 | 118.22 |
| Cavg | 59.50 | 38.30 | 37.34 | 64.12 | 63.29 | 4.96 | 18.52 | 55.16 |
| SD | 37.34 | 8.77 | 10.08 | 33.62 | 26.20 | 2.42 | 9.60 | 31.38 |
| NFNa | NFNH4 | NFK | NFMg | NFCa | ||
|---|---|---|---|---|---|---|
| Bulk Deposition | Average | 1.11 | 0.72 | 0.21 | 0.42 | 1.42 |
| SD | 0.95 | 0.20 | 0.25 | 0.29 | 0.60 | |
| Min | 0.12 | 0.47 | 0.03 | 0.11 | 0.72 | |
| Max | 2.82 | 1.19 | 0.78 | 1.02 | 2.91 | |
| Wet Deposition | Average | 0.95 | 0.90 | 0.07 | 0.27 | 0.82 |
| SD | 0.49 | 0.20 | 0.03 | 0.13 | 0.46 | |
| Min | 0.35 | 0.65 | 0.05 | 0.11 | 0.28 | |
| Max | 1.94 | 1.28 | 0.15 | 0.50 | 1.76 |
| Anion | β0 | β*Na βNa | β*NH4 βNH4 | β*K βK | β*Mg βMg | β*Ca βCa | R2 | |
|---|---|---|---|---|---|---|---|---|
| Bulk Deposition | [NO3−] | −0.339 | 1.915 * | −1.225 * | 0.353 | −0.006 | ||
| 15.667 * | −0.034 | 0.544 * | −0.435 * | 0.130 | −0.001 | 0.976 | ||
| [SO42−] | 0.074 | 0.746 | −0.195 | 0.449 | −0.083 | |||
| 2.540 | 0.019 | 0.548 | −0.178 | 0.427 | −0.042 | 0.963 | ||
| [Cl−] | 1.278 * | −0.375 | 0.063 | −0.031 | −0.067 | |||
| 36.155 | 1.457 * | −1.202 | 0.252 | −0.131 | −0.147 | 0.973 | ||
| Wet Deposition | −0.342 | 0.907 * | −0.045 | 0.536 | −0.049 | |||
| [NO3−] | 17.288 * | −0.089 | 0.304 * | −0.163 | 0.489 | −0.014 | 0.966 | |
| [SO42−] | −2.598 | 0.409 | 0.608 | 3.312 | −1.297 | |||
| 23.397 * | −0.779 | 0.157 | 2.534 | 3.475 | −0.417 | 0.908 | ||
| [Cl−] | −0.140 | 0.064 | 0.024 | 1.305 * | −0.522 * | |||
| 2.188 | −0.156 | 0.091 | 0.365 | 5.075 * | −0.621 * | 0.993 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gluščić, V.; Žužul, S.; Pehnec, G.; Jakovljević, I.; Smoljo, I.; Godec, R.; Bešlić, I.; Milinković, A.; Alempijević, S.B.; Frka, S. Sources, Ionic Composition and Acidic Properties of Bulk and Wet Atmospheric Deposition in the Eastern Middle Adriatic Region. Toxics 2023, 11, 551. https://doi.org/10.3390/toxics11070551
Gluščić V, Žužul S, Pehnec G, Jakovljević I, Smoljo I, Godec R, Bešlić I, Milinković A, Alempijević SB, Frka S. Sources, Ionic Composition and Acidic Properties of Bulk and Wet Atmospheric Deposition in the Eastern Middle Adriatic Region. Toxics. 2023; 11(7):551. https://doi.org/10.3390/toxics11070551
Chicago/Turabian StyleGluščić, Valentina, Silva Žužul, Gordana Pehnec, Ivana Jakovljević, Iva Smoljo, Ranka Godec, Ivan Bešlić, Andrea Milinković, Saranda Bakija Alempijević, and Sanja Frka. 2023. "Sources, Ionic Composition and Acidic Properties of Bulk and Wet Atmospheric Deposition in the Eastern Middle Adriatic Region" Toxics 11, no. 7: 551. https://doi.org/10.3390/toxics11070551
APA StyleGluščić, V., Žužul, S., Pehnec, G., Jakovljević, I., Smoljo, I., Godec, R., Bešlić, I., Milinković, A., Alempijević, S. B., & Frka, S. (2023). Sources, Ionic Composition and Acidic Properties of Bulk and Wet Atmospheric Deposition in the Eastern Middle Adriatic Region. Toxics, 11(7), 551. https://doi.org/10.3390/toxics11070551