
Oleanolic Acid Acetate Alleviates Cisplatin-Induced Nephrotoxicity via Inhibition of Apoptosis and Necroptosis In Vitro and In Vivo

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Viability
2.4. Flow Cytometric Analysis
2.5. Proteome Profiler Mouse Apoptosis Array
2.6. Animals
2.7. Mouse Model of Cisplatin-Induced Nephrotoxicity
2.8. Serum and Tissue Collection
2.9. Serum Analysis
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Histological Analysis
2.12. RNA Sequencing
2.13. Western Blot
2.14. Quantitative Polymerase Chain Reaction (qPCR)
2.15. Statistical Analysis
3. Results
3.1. OAA Suppressed Cisplatin-Induced Apoptosis In Vitro
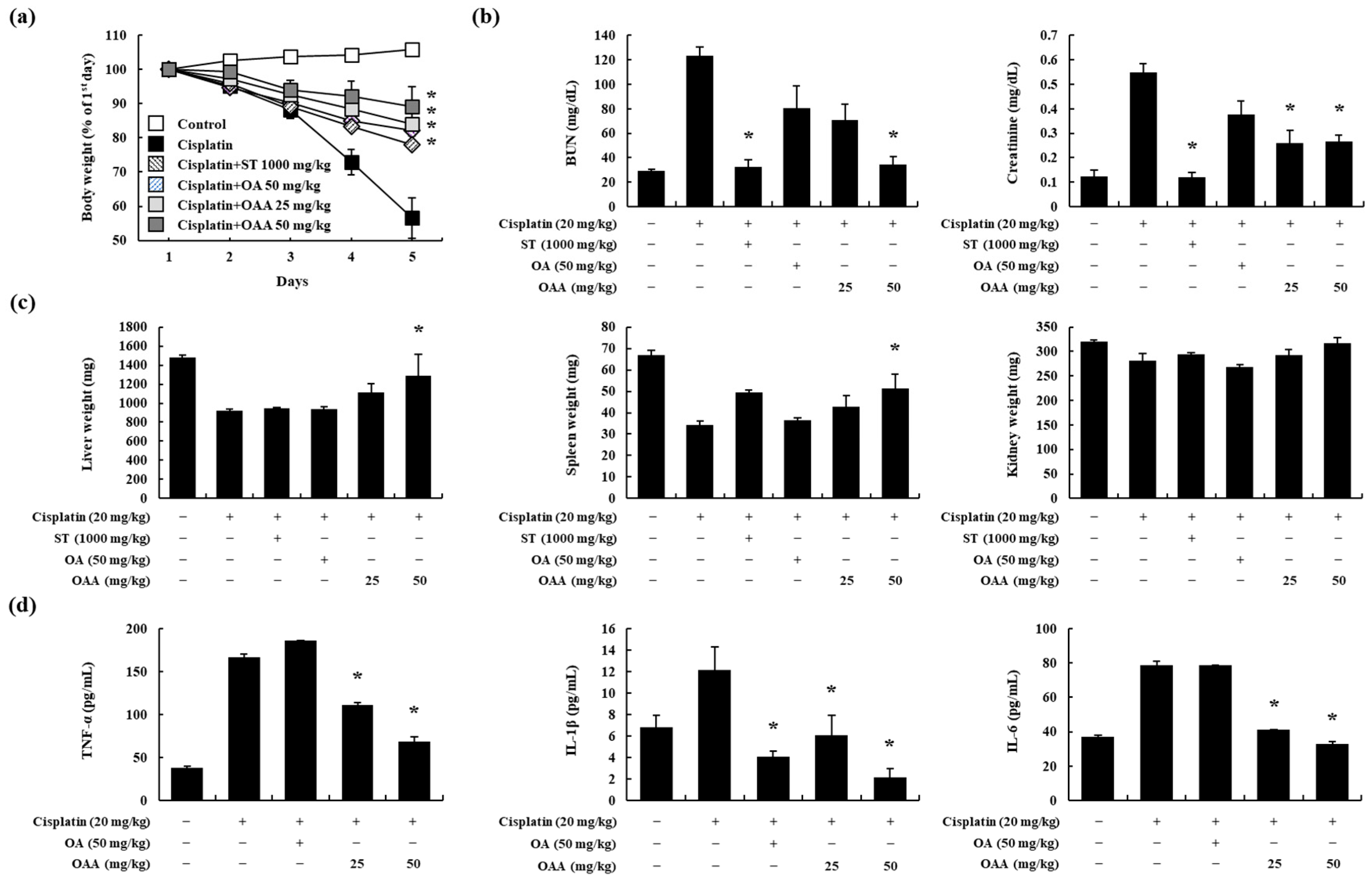
3.2. OAA Alleviated Cisplatin-Induced Kidney Injury In Vivo
3.3. OAA Regulated the Pattern of Gene Expression through RNA Sequencing In Vivo
3.4. OAA Inhibited Cisplatin-Induced Necroptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Hamroun, A.; Lenain, R.; Bigna, J.J.; Speyer, E.; Bui, L.; Chamley, P.; Pottier, N.; Cauffiez, C.; Dewaeles, E.; Dhalluin, X.; et al. Prevention of Cisplatin-Induced Acute Kidney Injury: A Systematic Review and Meta-Analysis. Drugs 2019, 79, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.; Wang, Q.; Ozkok, A.; Jani, A.; Li, H.; He, Z.; Ljubanovic, D.; Weiser-Evans, M.C.; Nemenoff, R.A.; Edelstein, C.L. CD4 T cell knockout does not protect against kidney injury and worsens cancer. J. Mol. Med. 2016, 94, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Roy, S.S.; Sk, U.H.; Bhattacharya, S. Amelioration of cisplatin-induced nephrotoxicity in mice by oral administration of diphenylmethyl selenocyanate. Free Radic. Res. 2011, 45, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Weisbord, S.D.; Palevsky, P.M. Design of Clinical Trials in Acute Kidney Injury: Lessons from the Past and Future Directions. Semin. Nephrol. 2016, 36, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef]
- Justice, A.E.; Karaderi, T.; Highland, H.M.; Young, K.L.; Graff, M.; Lu, Y.; Turcot, V.; Auer, P.L.; Fine, R.S.; Guo, X.; et al. Protein-coding variants implicate novel genes related to lipid homeostasis contributing to body-fat distribution. Nat. Genet. 2019, 51, 452–469. [Google Scholar] [CrossRef]
- Marakala, V. Neutrophil gelatinase-associated lipocalin (NGAL) in kidney injury—A systematic review. Clin. Chim. Acta. 2022, 536, 135–141. [Google Scholar] [CrossRef]
- Sieber, M.; Hoffmann, D.; Adler, M.; Vaidya, V.S.; Clement, M.; Bonventre, J.V.; Zidek, N.; Rached, E.; Amberg, A.; Callanan, J.J.; et al. Comparative analysis of novel noninvasive renal biomarkers and metabonomic changes in a rat model of gentamicin nephrotoxicity. Toxicol. Sci. 2009, 109, 336–349. [Google Scholar] [CrossRef]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Kadigamuwa, C.; Choksi, S.; Xu, Q.; Cataisson, C.; Greenbaum, S.S.; Yuspa, S.H.; Liu, Z.G. Role of Retinoic Acid Receptor-gamma in DNA Damage-Induced Necroptosis. iScience 2019, 17, 74–86. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 2003, 285, F610–F618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; An, Y.; Lu, X.; Zhong, H.; Zhu, Y.; Wu, Y.; Ma, F.; Yang, J.; Liu, Y.; Zhou, Z.; et al. A Novel Naphthalimide Compound Restores p53 Function in Non-small Cell Lung Cancer by Reorganizing the Bak.Bcl-xl Complex and Triggering Transcriptional Regulation. J. Biol. Chem. 2016, 291, 4211–4225. [Google Scholar] [CrossRef]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Ocker, M.; Hopfner, M. Apoptosis-modulating drugs for improved cancer therapy. Eur. Surg. Res. 2012, 48, 111–120. [Google Scholar] [CrossRef]
- Wang, D.H.; Hu, J.R.; Wang, L.Y.; Hu, Y.J.; Tan, F.Q.; Zhou, H.; Shao, J.Z.; Yang, W.X. The apoptotic function analysis of p53, Apaf1, Caspase3 and Caspase7 during the spermatogenesis of the Chinese fire-bellied newt Cynops orientalis. PLoS ONE 2012, 7, e39920. [Google Scholar] [CrossRef]
- Kirsch, D.G.; Doseff, A.; Chau, B.N.; Lim, D.S.; de Souza-Pinto, N.C.; Hansford, R.; Kastan, M.B.; Lazebnik, Y.A.; Hardwick, J.M. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 1999, 274, 21155–21161. [Google Scholar] [CrossRef]
- Erwig, L.P.; Henson, P.M. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008, 15, 243–250. [Google Scholar] [CrossRef]
- Tang, Q.; Li, W.; Dai, N.; Gao, Y.; Han, Y.; Cheng, G.; Gu, C. The Role of Necroptosis, Apoptosis, and Inflammation in Fowl Cholera-Associated Liver Injury in a Chicken Model. Avian. Dis. 2017, 61, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, Z.G. Execution of RIPK3-regulated necrosis. Mol. Cell Oncol. 2014, 1, e960759. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, H.B.; Fang, Y.L.; Zhang, Z.R.; Shao, J.; Hong, M.; Huang, C.J.; Liu, J.; Chen, R.Q. Cisplatin-induced necroptosis in TNFalpha dependent and independent pathways. Cell Signal. 2017, 31, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Dillon, C.P.; Weinlich, R.; Rodriguez, D.A.; Cripps, J.G.; Quarato, G.; Gurung, P.; Verbist, K.C.; Brewer, T.L.; Llambi, F.; Gong, Y.N.; et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Rickard, J.A.; O’Donnell, J.A.; Evans, J.M.; Lalaoui, N.; Poh, A.R.; Rogers, T.; Vince, J.E.; Lawlor, K.E.; Ninnis, R.L.; Anderton, H.; et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 2014, 157, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cao, Y.; Wang, H.X.; Zhao, L.; Chen, Y.X.; Zhong, K.H.; Li, G.W.; Wang, G.Q.; Huang, K.R.; Tong, A.P.; et al. Necrostatin-1 decreases necroptosis and inflammatory markers after intraventricular hemorrhage in mice. Neural Regen. Res. 2022, 17, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, X.; Zhu, Y.; Zhang, H.; Wang, H.; Ma, Q.; Yan, F.; Yang, Y.; Zhang, J.; Shi, H.; et al. The Caspase Inhibitor Z-VAD-FMK Alleviates Endotoxic Shock via Inducing Macrophages Necroptosis and Promoting MDSCs-Mediated Inhibition of Macrophages Activation. Front. Immunol. 2019, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Sato, S. Polyphenol-containing azuki bean (Vigna angularis) seed coats attenuate vascular oxidative stress and inflammation in spontaneously hypertensive rats. J. Nutr. Biochem. 2011, 22, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.M.; Lee, S.W.; Yun, B.R.; Hwang, B.S.; Kim, S.N.; Park, C.S.; Jeoung, S.H.; Kim, H.K.; Lee, W.S.; Rho, M.C. Vigna angularis inhibits IL-6-induced cellular signalling and ameliorates collagen-induced arthritis. Rheumatology 2014, 53, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; Kim, S.W.; Kim, D.S.; Lee, J.Y.; Lee, S.; Oh, H.M.; Ha, Y.S.; Yoo, J.; Park, P.H.; Shin, T.Y.; et al. Oleanolic acid acetate inhibits rheumatoid arthritis by modulating T cell immune responses and matrix-degrading enzymes. Toxicol. Appl. Pharmacol. 2016, 290, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dickey, D.T.; Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. Protection against cisplatin-induced toxicities by N-acetylcysteine and sodium thiosulfate as assessed at the molecular, cellular, and in vivo levels. J. Pharmacol. Exp. Ther. 2005, 314, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Jang, H.J.; Kim, M.H.; Lee, S.; Lee, S.W.; Lee, S.J.; Rho, M.C. Oleanolic Acid Acetate Exerts Anti-Inflammatory Activity via IKKalpha/beta Suppression in TLR3-Mediated NF-kappaB Activation. Molecules 2019, 24, 4002. [Google Scholar] [CrossRef] [PubMed]
- Rui, C.; Shi, S.N.; Ren, W.; Qin, X.; Zhuang, C.; Chen, X.; Chen, G.; Yu, J.; Wang, H.Y.; Cai, Z. The multitargeted kinase inhibitor KW-2449 ameliorates cisplatin-induced nephrotoxicity by targeting RIPK1-mediated necroptosis. Biochem. Pharmacol. 2021, 188, 114542. [Google Scholar] [CrossRef]
- Ye, L.; Pang, W.; Huang, Y.; Wu, H.; Huang, X.; Liu, J.; Wang, S.; Yang, C.; Pan, Q.; Liu, H. Lansoprazole promotes cisplatin-induced acute kidney injury via enhancing tubular necroptosis. J. Cell Mol. Med. 2021, 25, 2703–2713. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Wang, F.; Suo, X.G.; Gu, M.Z.; Wang, J.N.; Xu, C.H.; Dong, Y.H.; He, Y.; Zhang, Y.; Ji, M.L.; et al. Compound-42 alleviates acute kidney injury by targeting RIPK3-mediated necroptosis. Br. J. Pharmacol. 2023, 180, 2641–2660. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Faso, A.; Nishijima, T.F.; McGraw, K.A.; Galsky, M.D.; Milowsky, M.I. A Systematic Review of Strategies to Prevent Cisplatin-Induced Nephrotoxicity. Oncologist 2017, 22, 609–619. [Google Scholar] [CrossRef]
- Choi, J.K.; Oh, H.M.; Lee, S.; Park, J.W.; Khang, D.; Lee, S.W.; Lee, W.S.; Rho, M.C.; Kim, S.H. Oleanolic acid acetate inhibits atopic dermatitis and allergic contact dermatitis in a murine model. Toxicol. Appl. Pharmacol. 2013, 269, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Lee, S.; Kim, M.J.; Rho, M.C.; Jang, Y.H.; Kim, S.H. Oleanolic Acid Acetate Inhibits Mast Cell Activation in Ovalbumin-Induced Allergic Airway Inflammation. Allergy Asthma Immunol. Res. 2023, 15, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shu, B.; Tian, Y.; Wang, G.; Wang, Y.; Wang, J.; Dong, Y. Oleanolic acid induces osteosarcoma cell apoptosis by inhibition of Notch signaling. Mol. Carcinog. 2018, 57, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, C.; Li, X.; Liang, G. Oleanolic acid reduces oxidative stress and neuronal apoptosis after experimental subarachnoid hemorrhage by regulating Nrf2/HO-1 pathway. Drug Dev. Res. 2022, 83, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Tristao, V.R.; Pessoa, E.A.; Nakamichi, R.; Reis, L.A.; Batista, M.C.; Durao Junior Mde, S.; Monte, J.C. Synergistic effect of apoptosis and necroptosis inhibitors in cisplatin-induced nephrotoxicity. Apoptosis 2016, 21, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.R.; Mansouri, E.; Khodadadi, A. Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratisl. Lek. Listy 2017, 118, 123–128. [Google Scholar] [CrossRef]
- Yang, H.; Xu, S.; Tang, L.; Gong, J.; Fang, H.; Wei, J.; Su, D. Targeting of non-apoptotic cancer cell death mechanisms by quercetin: Implications in cancer therapy. Front. Pharmacol. 2022, 13, 1043056. [Google Scholar] [CrossRef]
- Khorsandi, L.; Saki, G.; Bavarsad, N.; Mombeini, M. Silymarin induces a multi-targeted cell death process in the human colon cancer cell line HT-29. Biomed. Pharmacother. 2017, 94, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Al-Salam, S.; Jagadeesh, G.S.; Sudhadevi, M.; Tageldeen, H.; Yasin, J. Galectin-3 Possesses Anti-Necroptotic and Anti-Apoptotic Effects in Cisplatin-Induced Acute Tubular Necrosis. Cell Physiol. Biochem. 2021, 55, 344–363. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Origin | Forward (5′–to–3′) | Reverse (5′–to–3′) |
|---|---|---|---|
| RIPK1 | Mouse | GAC TGT GTA CCC TTA CCT CCG A | CAC TGC GAT CAT TCT CGT CCT G |
| RIPK3 | Mouse | GAA GAC ACG GCA CTC CTT GGT A | CTT GAG GCA GTA GTT CTT GGT GG |
| MLKL | Mouse | CTG AGG GAA CTG CTG GAT AGA G | CGA GGA AAC TGG AGC TGC TGA T |
| LCN2 | Mouse | GGA CCA GGG CTG TCG CTA CT | GGT GGC CAC TTG CAT TGT |
| Bax | Mouse | TGG CAG CTG ACA TGT TTT CTG AC | TCA CCC AAC CAC CCT GGT CTT |
| Bcl-2 | Mouse | TCG CCC TGT GGA TGA CTG A | CAG AGA CAG CCA GGA GAA ATC |
| β-actin | Mouse | TAG ACT TCG AGC AGG AGA TG | TTG ATC TTC ATG GTG CTA GG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.; Kim, Y.-Y.; Jeong, S.; Lee, S.W.; Lee, S.-J.; Rho, M.-C.; Kim, S.-H.; Lee, S. Oleanolic Acid Acetate Alleviates Cisplatin-Induced Nephrotoxicity via Inhibition of Apoptosis and Necroptosis In Vitro and In Vivo. Toxics 2024, 12, 301. https://doi.org/10.3390/toxics12040301
Lee B, Kim Y-Y, Jeong S, Lee SW, Lee S-J, Rho M-C, Kim S-H, Lee S. Oleanolic Acid Acetate Alleviates Cisplatin-Induced Nephrotoxicity via Inhibition of Apoptosis and Necroptosis In Vitro and In Vivo. Toxics. 2024; 12(4):301. https://doi.org/10.3390/toxics12040301
Chicago/Turabian StyleLee, Bori, Yeon-Yong Kim, Seungwon Jeong, Seung Woong Lee, Seung-Jae Lee, Mun-Chual Rho, Sang-Hyun Kim, and Soyoung Lee. 2024. "Oleanolic Acid Acetate Alleviates Cisplatin-Induced Nephrotoxicity via Inhibition of Apoptosis and Necroptosis In Vitro and In Vivo" Toxics 12, no. 4: 301. https://doi.org/10.3390/toxics12040301
APA StyleLee, B., Kim, Y.-Y., Jeong, S., Lee, S. W., Lee, S.-J., Rho, M.-C., Kim, S.-H., & Lee, S. (2024). Oleanolic Acid Acetate Alleviates Cisplatin-Induced Nephrotoxicity via Inhibition of Apoptosis and Necroptosis In Vitro and In Vivo. Toxics, 12(4), 301. https://doi.org/10.3390/toxics12040301