Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder?

 ,
, 

Abstract
:1. Introduction
2. Neurobehavioral Effects of Developmental PCB Exposures
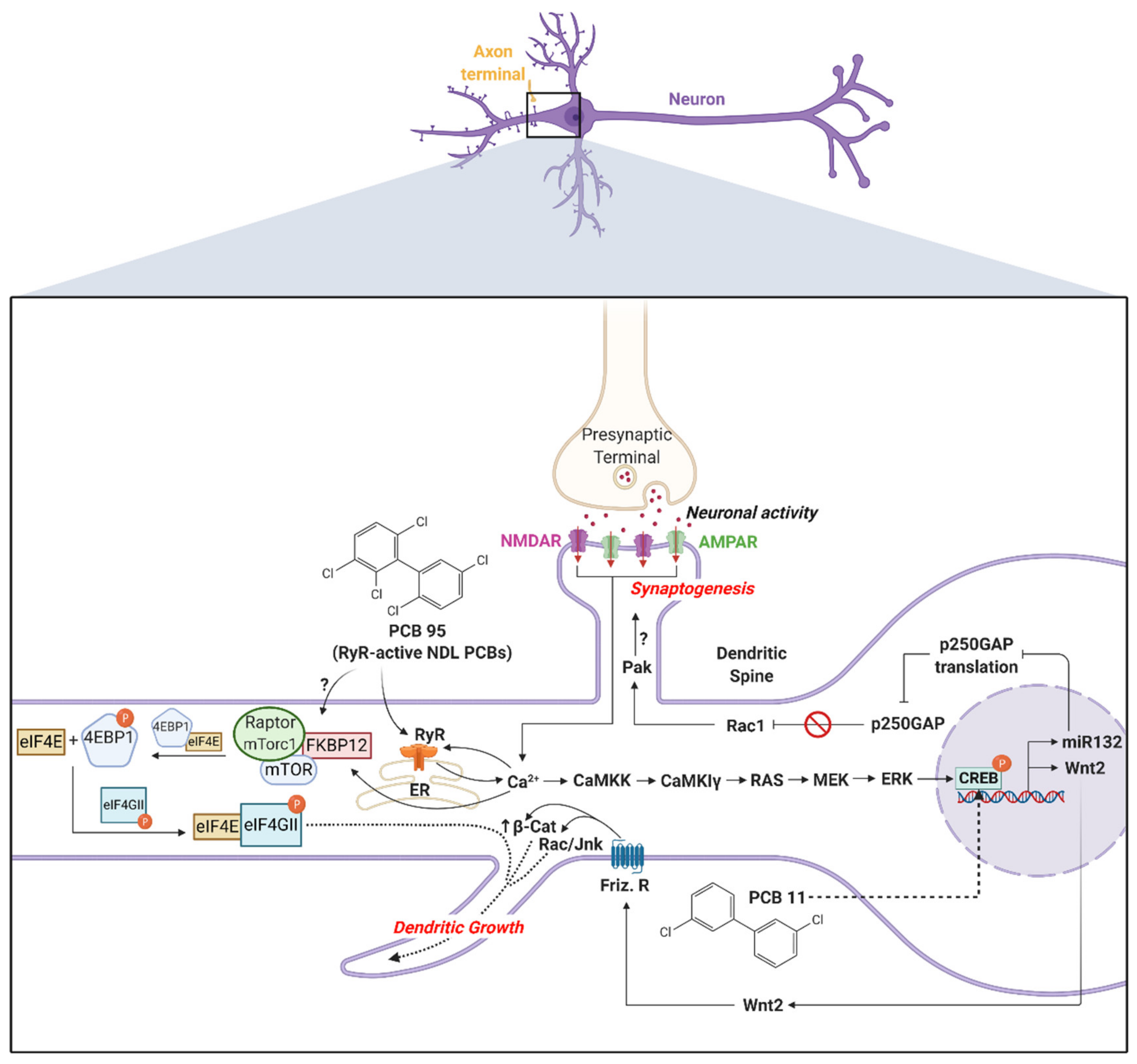
3. Mechanisms of PCB Developmental Neurotoxicity Relevant to ASD
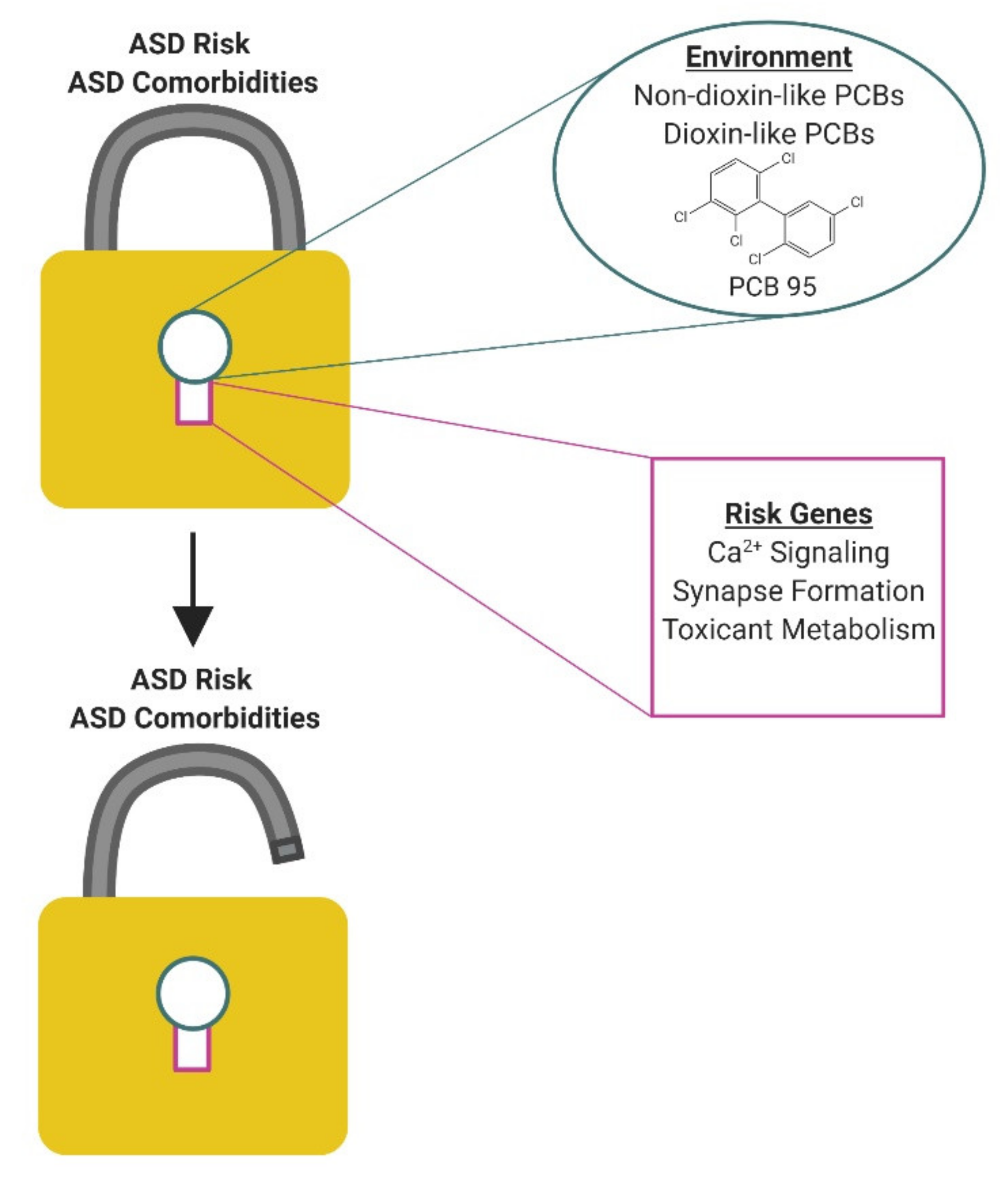
4. Mechanisms by Which PCBs Might Influence ASD Risk
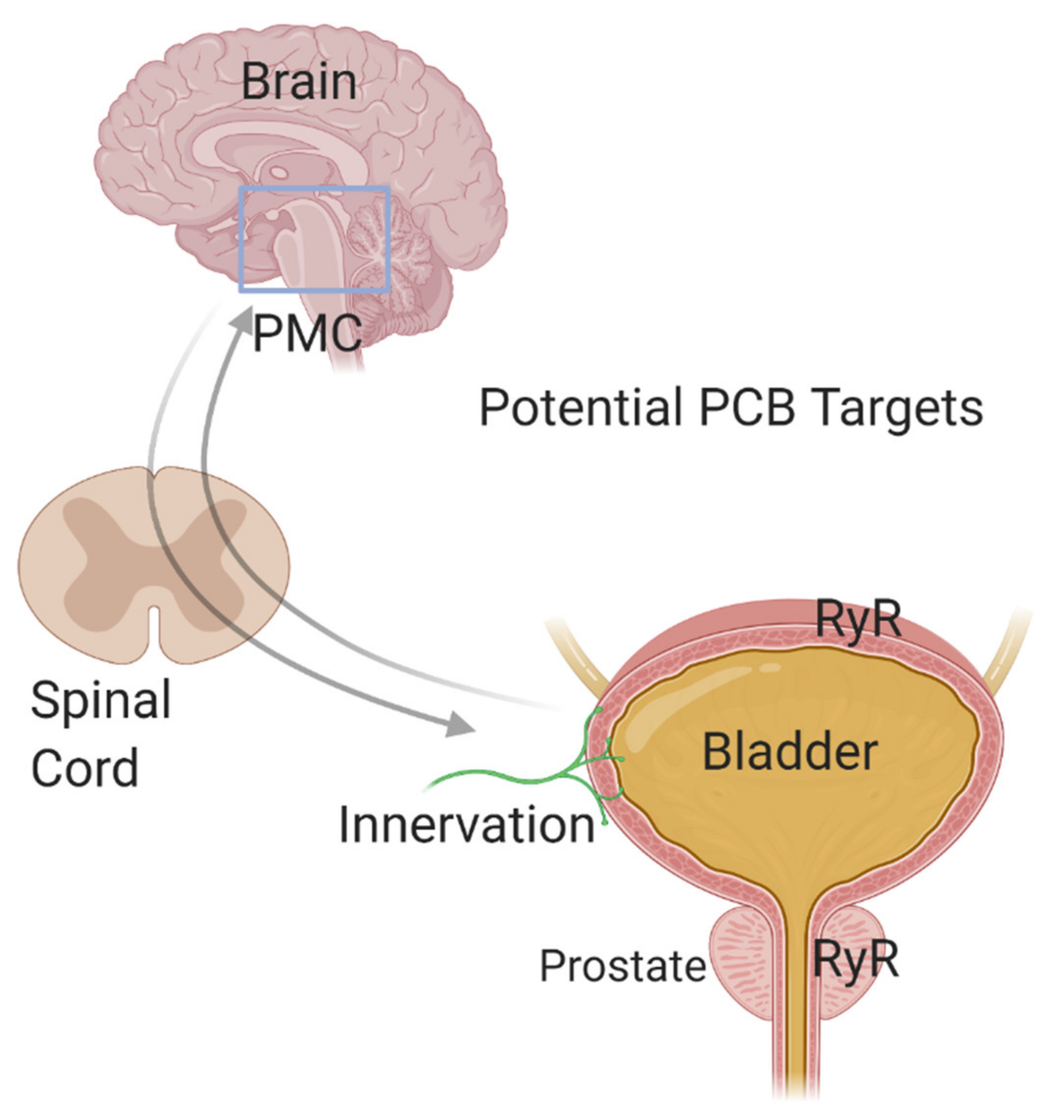
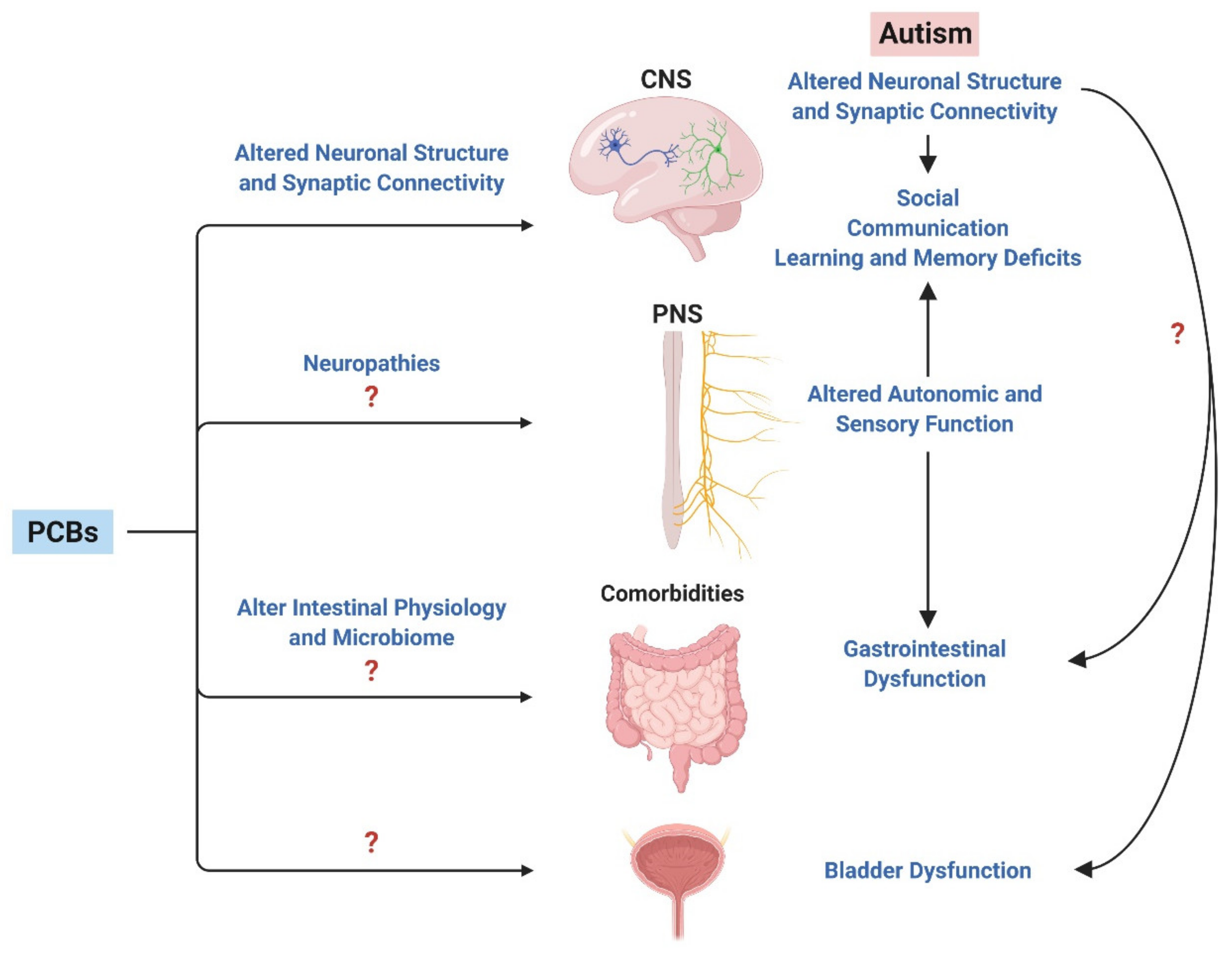
5. The Peripheral Nervous System: A Point of Convergence between ASD and PCBs?
6. A Role for PCBs in ASD Comorbidities?
7. Conclusions and the Path Forward
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Doshi-Velez, F.; Ge, Y.; Kohane, I. Comorbidity clusters in autism spectrum disorders: An electronic health record time-series analysis. Pediatrics 2014, 133, e54–e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazeer, A.; Ghaziuddin, M. Autism spectrum disorders: Clinical features and diagnosis. Pediatric Clin. N. Am. 2012, 59, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Lu, Y.; Li, Y.; Shi, J.; Cui, H.; Gu, Y.; Li, Y.; Zhong, W.; Zhu, X.; Liu, Y.; et al. Prevalence of autism spectrum disorder in Asia: A systematic review and meta-analysis. Psychiatry Res. 2020, 284, 112679. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Leventhal, B.L.; Koh, Y.J.; Fombonne, E.; Laska, E.; Lim, E.C.; Cheon, K.A.; Kim, S.J.; Kim, Y.K.; Lee, H.; et al. Prevalence of autism spectrum disorders in a total population sample. Am. J. Psychiatry 2011, 168, 904–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumberg, S.J.; Bramlett, M.D.; Kogan, M.D.; Schieve, L.A.; Jones, J.R.; Lu, M.C. Changes in prevalence of parent-reported autism spectrum disorder in school-aged U.S. children: 2007 to 2011–2012. Natl. Health Stat. Rep. 2013, 65, 1–11. [Google Scholar]
- Leibson, C.; Weaver, A.; Myers, S.; Long, K.; Ransom, J.; Voigt, R.; Katusic, S. Objective Estimates of Direct-Medical Costs among Persons Aged 3 to 38 Years with and without Research-Defined Autism Spectrum Disorder Ascertained During Childhood: A Population-Based Birth-Cohort Study. Value Health 2020, 23, 595–605. [Google Scholar] [CrossRef]
- Hong, M.; Lee, S.M.; Park, S.; Yoon, S.J.; Kim, Y.E.; Oh, I.H. Prevalence and Economic Burden of Autism Spectrum Disorder in South Korea Using National Health Insurance Data from 2008 to 2015. J. Autism Dev. Disord. 2020, 50, 333–339. [Google Scholar] [CrossRef]
- Schofield, D.; Zeppel, M.J.B.; Tanton, R.; Veerman, J.L.; Kelly, S.J.; Passey, M.E.; Shrestha, R.N. Intellectual disability and autism: Socioeconomic impacts of informal caring, projected to 2030. Br. J. Psychiatry 2019. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Hof, P.R. The emerging neuroscience of autism spectrum disorders. Brain Res. 2011, 1380, 1–2. [Google Scholar] [CrossRef]
- El-Fishawy, P.; State, M.W. The genetics of autism: Key issues, recent findings, and clinical implications. Pediatric Clin. N. Am. 2010, 33, 83–105. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, D.H. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delorme, R.; Ey, E.; Toro, R.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Progress toward treatments for synaptic defects in autism. Nat. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrigan, P.J.; Lambertini, L.; Birnbaum, L.S. A research strategy to discover the environmental causes of autism and neurodevelopmental disabilities. Environ. Health Perspect. 2012, 120, a258–a260. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Aldinger, K.A.; Levitt, P. Modeling of autism genetic variations in mice: Focusing on synaptic and microcircuit dysfunctions. Dev. Neurosci. 2012, 34, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.R. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr. Opin. Neurol. 2010, 23, 103–110. [Google Scholar] [CrossRef]
- Levitt, P.; Campbell, D.B. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J. Clin. Investig. 2009, 119, 747–754. [Google Scholar] [CrossRef]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Grether, J.K.; Rosen, N.J.; Smith, K.S.; Croen, L.A. Investigation of shifts in autism reporting in the California Department of Developmental Services. J. Autism Dev. Disord. 2009, 39, 1412–1419. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Delwiche, L. The rise in autism and the role of age at diagnosis. Epidemiology 2009, 20, 84–90. [Google Scholar] [CrossRef] [Green Version]
- King, M.; Bearman, P. Diagnostic change and the increased prevalence of autism. Int. J. Epidemiol. 2009, 38, 1224–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Bolte, S.; Girdler, S.; Marschik, P.B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol. Life Sci. 2019, 76, 1275–1297. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, T.; Molander, F.; Taylor, M.J.; Jonsson, U.; Bolte, S. Early environmental risk factors for neurodevelopmental disorders—A systematic review of twin and sibling studies. Dev. Psychopathol. 2020, 1–48. [Google Scholar] [CrossRef]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Landrigan, P.J. What causes autism? Exploring the environmental contribution. Curr. Opin. Pediatrics 2010, 22, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Stamou, M.; Streifel, K.M.; Goines, P.E.; Lein, P.J. Neuronal connectivity as a convergent target of gene x environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol. Teratol. 2013, 36, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Bill, B.R.; Geschwind, D.H. Genetic advances in autism: Heterogeneity and convergence on shared pathways. Curr. Opin. Genet. Dev. 2009, 19, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Hu, V.W. From genes to environment: Using integrative genomics to build a “systems-level” understanding of autism spectrum disorders. Child. Dev. 2013, 84, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Visser, J.C.; Rommelse, N.; Vink, L.; Schrieken, M.; Oosterling, I.J.; van der Gaag, R.J.; Buitelaar, J.K. Narrowly versus broadly defined autism spectrum disorders: Differences in pre- and perinatal risk factors. J. Autism Dev. Disord. 2013, 43, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Pessah, I.N.; Lein, P.J. Evidence for environmental susceptibility in autism: What we need to know about gene x environment interactions. In Autism: Current Theories and Evidence; Zimmerman, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 409–428. [Google Scholar]
- Lein, P.J. Overview of the role of environmental factors in neurodevelopmental disorders. In Environmental Factors in Neurodevelopmental and Neurodegenerative Disorders; Costa, L.G., Aschner, M., Eds.; Elsevier, Inc.: Oxford, UK, 2015; pp. 3–20. [Google Scholar]
- Grimm, F.A.; Hu, D.; Kania-Korwel, I.; Lehmler, H.J.; Ludewig, G.; Hornbuckle, K.C.; Duffel, M.W.; Bergman, A.; Robertson, L.W. Metabolism and metabolites of polychlorinated biphenyls. Crit. Rev. Toxicol. 2015, 45, 245–272. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.; Birnbaum, L.; Bosveld, A.T.; Brunstrom, B.; Cook, P.; Feeley, M.; Giesy, J.P.; Hanberg, A.; Hasegawa, R.; Kennedy, S.W.; et al. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 1998, 106, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.X.; Hornbuckle, K.C.; Thorne, P.S. Human Serum from Urban and Rural Adolescents and Their Mothers Shows Exposure to Polychlorinated Biphenyls Not Found in Commercial Mixtures. Environ. Sci. Technol. 2015, 49, 8105–8112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, S.; Morgan, R.K.; Feng, W.; Lin, Y.; Li, X.; Luna, C.; Koch, M.; Bansal, R.; Duffel, M.W.; Puschner, B.; et al. Comparative Analyses of the 12 Most Abundant PCB Congeners Detected in Human Maternal Serum for Activity at the Thyroid Hormone Receptor and Ryanodine Receptor. Environ. Sci. Technol. 2019, 53, 3948–3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Hornbuckle, K.C. Inadvertent polychlorinated biphenyls in commercial paint pigments. Environ. Sci. Technol. 2010, 44, 2822–2827. [Google Scholar] [CrossRef]
- Sethi, S.; Chen, X.; Kass, P.H.; Puschner, B. Polychlorinated biphenyl and polybrominated diphenyl ether profiles in serum from cattle, sheep, and goats across California. Chemosphere 2017, 181, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.; Xue, J.; Williams, R.; Jones, P.; Whitaker, D. Polychlorinated Biphenyls (PCBs) in School Buildings: Sources, Environmental Levels, and Exposures; United States Environmental Protection Agency: Washington, DC, USA, 2012; p. 150. [Google Scholar]
- Ampleman, M.D.; Martinez, A.; DeWall, J.; Rawn, D.F.; Hornbuckle, K.C.; Thorne, P.S. Inhalation and dietary exposure to PCBs in urban and rural cohorts via congener-specific measurements. Environ. Sci. Technol. 2015, 49, 1156–1164. [Google Scholar] [CrossRef]
- Dewailly, E.; Mulvad, G.; Pedersen, H.S.; Ayotte, P.; Demers, A.; Weber, J.P.; Hansen, J.C. Concentration of organochlorines in human brain, liver, and adipose tissue autopsy samples from Greenland. Environ. Health Perspect. 1999, 107, 823–828. [Google Scholar] [CrossRef]
- Chu, S.; Covaci, A.; Schepens, P. Levels and chiral signatures of persistent organochlorine pollutants in human tissues from Belgium. Environ. Res. 2003, 93, 167–176. [Google Scholar] [CrossRef]
- Mitchell, M.M.; Woods, R.; Chi, L.H.; Schmidt, R.J.; Pessah, I.N.; Kostyniak, P.J.; LaSalle, J.M. Levels of select PCB and PBDE congeners in human postmortem brain reveal possible environmental involvement in 15q11-q13 duplication autism spectrum disorder. Environ. Mol. Mutagenesis 2012, 53, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Pessah, I.N.; Lein, P.J.; Seegal, R.F.; Sagiv, S.K. Neurotoxicity of polychlorinated biphenyls and related organohalogens. Acta Neuropathol. 2019, 138, 363–387. [Google Scholar] [CrossRef]
- Thompson, M.R.; Boekelheide, K. Multiple environmental chemical exposures to lead, mercury and polychlorinated biphenyls among childbearing-aged women (NHANES 1999–2004): Body burden and risk factors. Environ. Res. 2013, 121, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Koh, W.X.; Hornbuckle, K.C.; Marek, R.F.; Wang, K.; Thorne, P.S. Hydroxylated polychlorinated biphenyls in human sera from adolescents and their mothers living in two U.S. Midwestern communities. Chemosphere 2016, 147, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Marek, R.F.; Thorne, P.S.; Wang, K.; Dewall, J.; Hornbuckle, K.C. PCBs and OH-PCBs in serum from children and mothers in urban and rural U.S. communities. Environ. Sci. Technol. 2013, 47, 3353–3361. [Google Scholar] [CrossRef]
- Sethi, S.; Keil, K.P.; Chen, H.; Hayakawa, K.; Li, X.; Lin, Y.; Lehmler, H.J.; Puschner, B.; Lein, P.J. Detection of 3,3’-Dichlorobiphenyl in Human Maternal Plasma and Its Effects on Axonal and Dendritic Growth in Primary Rat Neurons. Toxicol. Sci. 2017, 158, 401–411. [Google Scholar] [CrossRef]
- Eskenazi, B.; Rauch, S.A.; Tenerelli, R.; Huen, K.; Holland, N.T.; Lustig, R.H.; Kogut, K.; Bradman, A.; Sjodin, A.; Harley, K.G. In utero and childhood DDT, DDE, PBDE and PCBs exposure and sex hormones in adolescent boys: The CHAMACOS study. Int. J. Hyg. Environ. Health 2017, 220, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Granillo, L.; Sethi, S.; Keil, K.P.; Lin, Y.; Ozonoff, S.; Iosif, A.M.; Puschner, B.; Schmidt, R.J. Polychlorinated biphenyls influence on autism spectrum disorder risk in the MARBLES cohort. Environ. Res. 2019, 171, 177–184. [Google Scholar] [CrossRef]
- Baba, T.; Ito, S.; Yuasa, M.; Yoshioka, E.; Miyashita, C.; Araki, A.; Sasaki, S.; Kobayashi, S.; Kajiwara, J.; Hori, T.; et al. Association of prenatal exposure to PCDD/Fs and PCBs with maternal and infant thyroid hormones: The Hokkaido Study on Environment and Children’s Health. Sci. Total Environ. 2018, 615, 1239–1246. [Google Scholar] [CrossRef] [Green Version]
- Hisada, A.; Shimodaira, K.; Okai, T.; Watanabe, K.; Takemori, H.; Takasuga, T.; Koyama, M.; Watanabe, N.; Suzuki, E.; Shirakawa, M.; et al. Associations between levels of hydroxylated PCBs and PCBs in serum of pregnant women and blood thyroid hormone levels and body size of neonates. Int. J. Hyg. Environ. Health 2014, 217, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, A.H.; Hoyer, B.B.; Julvez, J.; Sunyer, J.; Pedersen, H.S.; Lenters, V.; Jonsson, B.A.G.; Bonde, J.P.; Toft, G. Prenatal and Postnatal PCB-153 and p,p’-DDE Exposures and Behavior Scores at 5–9 Years of Age among Children in Greenland and Ukraine. Environ. Health Perspect. 2017, 125, 107002. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, M.H.; Samms-Vaughan, M.; Hessabi, M.; Dickerson, A.S.; Lee, M.; Bressler, J.; Tomechko, S.E.; Moreno, E.K.; Loveland, K.A.; Desai, C.C.; et al. Concentrations of Polychlorinated Biphenyls and Organochlorine Pesticides in Umbilical Cord Blood Serum of Newborns in Kingston, Jamaica. Int. J. Environ. Res. Public Health 2016, 13, 1032. [Google Scholar] [CrossRef] [Green Version]
- Lanting, C.I.; Huisman, M.; Muskiet, F.A.; van der Paauw, C.G.; Essed, C.E.; Boersma, E.R. Polychlorinated biphenyls in adipose tissue, liver, and brain from nine stillborns of varying gestational ages. Pediatric Res. 1998, 44, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, C.; Uchi, H.; Tsukimori, K.; Yamada, H.; Akahane, M.; Imamura, T.; Utani, A.; Furue, M. Yusho and its latest findings-A review in studies conducted by the Yusho Group. Environ. Int. 2015, 82, 41–48. [Google Scholar] [CrossRef]
- Hsu, S.T.; Ma, C.I.; Hsu, S.K.; Wu, S.S.; Hsu, N.H.; Yeh, C.C.; Wu, S.B. Discovery and epidemiology of PCB poisoning in Taiwan: A four-year followup. Environ. Health Perspect. 1985, 59, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, S.A.; Bos, A.F.; Sauer, P.J.; Roze, E. Developmental neurotoxicity of persistent organic pollutants: An update on childhood outcome. Arch. Toxicol. 2015, 89, 687–709. [Google Scholar] [CrossRef]
- Korrick, S.A.; Sagiv, S.K. Polychlorinated biphenyls, organochlorine pesticides and neurodevelopment. Curr. Opin. Pediatrics 2008, 20, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, S.K.; Thurston, S.W.; Bellinger, D.C.; Tolbert, P.E.; Altshul, L.M.; Korrick, S.A. Prenatal organochlorine exposure and behaviors associated with attention deficit hyperactivity disorder in school-aged children. Am. J. Epidemiol. 2010, 171, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Schantz, S.L.; Widholm, J.J.; Rice, D.C. Effects of PCB exposure on neuropsychological function in children. Environ. Health Perspect. 2003, 111, 357–576. [Google Scholar] [CrossRef] [Green Version]
- Klocke, C.; Sethi, S.; Lein, P.J. The developmental neurotoxicity of legacy vs. contemporary polychlorinated biphenyls (PCBs): Similarities and differences. Environ. Sci. Pollut. Res. Int. 2020, 27, 8885–8896. [Google Scholar] [CrossRef]
- Nowack, N.; Wittsiepe, J.; Kasper-Sonnenberg, M.; Wilhelm, M.; Scholmerich, A. Influence of Low-Level Prenatal Exposure to PCDD/Fs and PCBs on Empathizing, Systemizing and Autistic Traits: Results from the Duisburg Birth Cohort Study. PLoS ONE 2015, 10, e0129906. [Google Scholar] [CrossRef]
- Braun, J.M.; Kalkbrenner, A.E.; Just, A.C.; Yolton, K.; Calafat, A.M.; Sjodin, A.; Hauser, R.; Webster, G.M.; Chen, A.; Lanphear, B.P. Gestational exposure to endocrine-disrupting chemicals and reciprocal social, repetitive, and stereotypic behaviors in 4- and 5-year-old children: The HOME study. Environ. Health Perspect. 2014, 122, 513–520. [Google Scholar] [CrossRef]
- Bach, M.A.; Samms-Vaughan, M.; Hessabi, M.; Bressler, J.; Lee, M.; Zhang, J.; Shakespeare-Pellington, S.; Grove, M.L.; Loveland, K.A.; Rahbar, M.H. Association of Polychlorinated Biphenyls and Organochlorine Pesticides with Autism Spectrum Disorder in Jamaican Children. Res. Autism Spectr. Disord. 2020, 76, 101587. [Google Scholar] [CrossRef] [PubMed]
- Lyall, K.; Croen, L.A.; Sjodin, A.; Yoshida, C.K.; Zerbo, O.; Kharrazi, M.; Windham, G.C. Polychlorinated Biphenyl and Organochlorine Pesticide Concentrations in Maternal Mid-Pregnancy Serum Samples: Association with Autism Spectrum Disorder and Intellectual Disability. Environ. Health Perspect. 2017, 125, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, B.A.; Lanphear, B.P.; Venners, S.A.; Arbuckle, T.E.; Braun, J.M.; Muckle, G.; Fraser, W.D.; McCandless, L.C. Assessing the Relation between Plasma PCB Concentrations and Elevated Autistic Behaviours using Bayesian Predictive Odds Ratios. Int. J. Environ. Res. Public Health 2019, 16, 457. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.C.; Krishnan, K.; Reilly, M.P. Endocrine-disrupting chemicals: Effects on neuroendocrine systems and the neurobiology of social behavior. Horm. Behav. 2019, 111, 7–22. [Google Scholar] [CrossRef]
- Sable, H.J.K.; Schantz, S.L. Executive Function Following Developmental Exposure to Polychlorinated Biphenyls (PCBs): What Animal Models Have Told Us; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 147–167. [Google Scholar]
- Ulbrich, B.; Stahlmann, R. Developmental toxicity of polychlorinated biphenyls (PCBs): A systematic review of experimental data. Arch. Toxicol. 2004, 78, 252–268. [Google Scholar] [CrossRef]
- Winneke, G. Developmental aspects of environmental neurotoxicology: Lessons from lead and polychlorinated biphenyls. J. Neurol. Sci. 2011, 308, 9–15. [Google Scholar] [CrossRef]
- Klocke, C.; Lein, P.J. Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 1013. [Google Scholar] [CrossRef] [Green Version]
- Pessah, I.N.; Cherednichenko, G.; Lein, P.J. Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol. Ther. 2010, 125, 260–285. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Mohrle, D.; Fernandez, M.; Penagarikano, O.; Frick, A.; Allman, B.; Schmid, S. What we can learn from a genetic rodent model about autism. Neurosci. Biobehav. Rev. 2020, 109, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Silverman, J.L.; Crawley, J.N. Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci. 2011, 56, 8–26. [Google Scholar] [CrossRef]
- Crawley, J.N. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007, 17, 448–459. [Google Scholar] [CrossRef]
- Reilly, M.P.; Weeks, C.D.; Crews, D.; Gore, A.C. Application of a novel social choice paradigm to assess effects of prenatal endocrine-disrupting chemical exposure in rats (Rattus norvegicus). J. Comp. Psychol. (Wash. D.C. 1983) 2018, 132, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.A.; Shair, H.N.; Brunelli, S.A. Ultrasonic vocalizations in rat and mouse pups. Curr. Protoc. Neurosci. 2002, 17, 8–14. [Google Scholar] [CrossRef]
- Winslow, J.T. Mouse social recognition and preference. Curr. Protoc. Neurosci. 2003, 22, 8–16. [Google Scholar] [CrossRef]
- Karkaba, A.; Soualeh, N.; Soulimani, R.; Bouayed, J. Perinatal effects of exposure to PCBs on social preferences in young adult and middle-aged offspring mice. Horm. Behav. 2017, 96, 137–146. [Google Scholar] [CrossRef]
- Bell, M.R.; Thompson, L.M.; Rodriguez, K.; Gore, A.C. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 1. Sexually dimorphic effects on social and anxiety-like behaviors. Horm. Behav. 2016, 78, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolous-Jamshidi, B.; Cromwell, H.C.; McFarland, A.M.; Meserve, L.A. Perinatal exposure to polychlorinated biphenyls alters social behaviors in rats. Toxicol. Lett. 2010, 199, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.P.; Weeks, C.D.; Topper, V.Y.; Thompson, L.M.; Crews, D.; Gore, A.C. The effects of prenatal PCBs on adult social behavior in rats. Horm. Behav. 2015, 73, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topper, V.Y.; Reilly, M.P.; Wagner, L.M.; Thompson, L.M.; Gillette, R.; Crews, D.; Gore, A.C. Social and neuromolecular phenotypes are programmed by prenatal exposures to endocrine-disrupting chemicals. Mol. Cell Endocrinol. 2019, 479, 133–146. [Google Scholar] [CrossRef]
- Uwimana, E.; Ruiz, P.; Li, X.; Lehmler, H.J. Human CYP2A6, CYP2B6, AND CYP2E1 Atropselectively Metabolize Polychlorinated Biphenyls to Hydroxylated Metabolites. Environ. Sci. Technol. 2019, 53, 2114–2123. [Google Scholar] [CrossRef]
- Tehrani, R.; Van Aken, B. Hydroxylated polychlorinated biphenyls in the environment: Sources, fate, and toxicities. Environ. Sci. Pollut. Res. Int. 2014, 21, 6334–6345. [Google Scholar] [CrossRef]
- Ma, S.; Ren, G.; Zeng, X.; Yu, Z.; Sheng, G.; Fu, J. Polychlorinated biphenyls and their hydroxylated metabolites in the serum of e-waste dismantling workers from eastern China. Environ. Geochem. Health 2018, 40, 1931–1940. [Google Scholar] [CrossRef]
- Berghuis, S.A.; Van Braeckel, K.; Sauer, P.J.J.; Bos, A.F. Prenatal exposure to persistent organic pollutants and cognition and motor performance in adolescence. Environ. Int. 2018, 121, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, S.A.; Soechitram, S.D.; Hitzert, M.M.; Sauer, P.J.; Bos, A.F. Prenatal exposure to polychlorinated biphenyls and their hydroxylated metabolites is associated with motor development of three-month-old infants. Neurotoxicology 2013, 38, 124–130. [Google Scholar] [CrossRef]
- Berghuis, S.A.; Soechitram, S.D.; Sauer, P.J.; Bos, A.F. Prenatal exposure to polychlorinated biphenyls and their hydroxylated metabolites is associated with neurological functioning in 3-month-old infants. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 142, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.Y.; Park, J.S.; Sovcikova, E.; Kocan, A.; Linderholm, L.; Bergman, A.; Trnovec, T.; Hertz-Picciotto, I. Exposure to hydroxylated polychlorinated biphenyls (OH-PCBs) in the prenatal period and subsequent neurodevelopment in eastern Slovakia. Environ. Health Perspect. 2009, 117, 1600–1606. [Google Scholar] [CrossRef]
- Ruel, M.V.M.; Bos, A.F.; Soechitram, S.D.; Meijer, L.; Sauer, P.J.J.; Berghuis, S.A. Prenatal exposure to organohalogen compounds and children’s mental and motor development at 18 and 30 months of age. Neurotoxicology 2019, 72, 6–14. [Google Scholar] [CrossRef]
- Takeuchi, S.; Shiraishi, F.; Kitamura, S.; Kuroki, H.; Jin, K.; Kojima, H. Characterization of steroid hormone receptor activities in 100 hydroxylated polychlorinated biphenyls, including congeners identified in humans. Toxicology 2011, 289, 112–121. [Google Scholar] [CrossRef]
- Pencikova, K.; Svrzkova, L.; Strapacova, S.; Neca, J.; Bartonkova, I.; Dvorak, Z.; Hyzdalova, M.; Pivnicka, J.; Palkova, L.; Lehmler, H.J.; et al. In vitro profiling of toxic effects of prominent environmental lower-chlorinated PCB congeners linked with endocrine disruption and tumor promotion. Environ. Pollut. 2018, 237, 473–486. [Google Scholar] [CrossRef]
- Itoh, S.; Baba, T.; Yuasa, M.; Miyashita, C.; Kobayashi, S.; Araki, A.; Sasaki, S.; Kajiwara, J.; Hori, T.; Todaka, T.; et al. Association of maternal serum concentration of hydroxylated polychlorinated biphenyls with maternal and neonatal thyroid hormones: The Hokkaido birth cohort study. Environ. Res. 2018, 167, 583–590. [Google Scholar] [CrossRef]
- Kania-Korwel, I.; Barnhart, C.D.; Stamou, M.; Truong, K.M.; El-Komy, M.H.; Lein, P.J.; Veng-Pedersen, P.; Lehmler, H.J. 2,2’,3,5’,6-Pentachlorobiphenyl (PCB 95) and its hydroxylated metabolites are enantiomerically enriched in female mice. Environ. Sci. Technol. 2012, 46, 11393–11401. [Google Scholar] [CrossRef] [Green Version]
- Kania-Korwel, I.; Barnhart, C.D.; Lein, P.J.; Lehmler, H.J. Effect of pregnancy on the disposition of 2,2’,3,5’,6-pentachlorobiphenyl (PCB 95) atropisomers and their hydroxylated metabolites in female mice. Chem. Res. Toxicol. 2015, 28, 1774–1783. [Google Scholar] [CrossRef] [Green Version]
- Bourgeron, T. A synaptic trek to autism. Curr. Opin. Neurobiol. 2009, 19, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasek, R. Studying the intrinsic determinants of neuronal form and function. In Intrinsic Determinants of Neuronal Form and Function; Lasek, R., Black, M.B., Eds.; A.R. Liss: New York, NY, USA, 1988; pp. 3–58. [Google Scholar]
- Berger-Sweeney, J.; Hohmann, C.F. Behavioral consequences of abnormal cortical development: Insights into developmental disabilities. Behav. Brain Res. 1997, 86, 121–142. [Google Scholar] [CrossRef]
- Cremer, H.; Chazal, G.; Goridis, C.; Represa, A. NCAM is essential for axonal growth and fasciculation in the hippocampus. Mol. Cell Neurosci. 1997, 8, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Maier, D.L.; Mani, S.; Donovan, S.L.; Soppet, D.; Tessarollo, L.; McCasland, J.S.; Meiri, K.F. Disrupted cortical map and absence of cortical barrels in growth-associated protein (GAP)-43 knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 9397–9402. [Google Scholar] [CrossRef] [Green Version]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. S3), 511–533. [Google Scholar] [CrossRef]
- Copf, T. Impairments in dendrite morphogenesis as etiology for neurodevelopmental disorders and implications for therapeutic treatments. Neurosci. Biobehav. Rev. 2016, 68, 946–978. [Google Scholar] [CrossRef]
- Engle, E.C. Human genetic disorders of axon guidance. Cold Spring Harb. Perspect. Biol. 2010, 2, a001784. [Google Scholar] [CrossRef]
- Martinez-Cerdeno, V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 2017, 77, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, M.A.; Cowan, C.W. Signaling mechanisms of axon guidance and early synaptogenesis. Curr. Top. Behav. Neurosci. 2014, 16, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Supekar, K.; Uddin, L.Q.; Khouzam, A.; Phillips, J.; Gaillard, W.D.; Kenworthy, L.E.; Yerys, B.E.; Vaidya, C.J.; Menon, V. Brain hyperconnectivity in children with autism and its links to social deficits. Cell Rep. 2013, 5, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, D.L.; Meserve, L.A.; Bingman, V.P. Reduced growth of intra- and infra-pyramidal mossy fibers is produced by continuous exposure to polychlorinated biphenyl. Toxicology 1999, 138, 11–17. [Google Scholar] [CrossRef]
- Lein, P.J.; Yang, D.; Bachstetter, A.D.; Tilson, H.A.; Harry, G.J.; Mervis, R.F.; Kodavanti, P.R. Ontogenetic alterations in molecular and structural correlates of dendritic growth after developmental exposure to polychlorinated biphenyls. Environ. Health Perspect. 2007, 115, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Roegge, C.S.; Morris, J.R.; Villareal, S.; Wang, V.C.; Powers, B.E.; Klintsova, A.Y.; Greenough, W.T.; Pessah, I.N.; Schantz, S.L. Purkinje cell and cerebellar effects following developmental exposure to PCBs and/or MeHg. Neurotoxicol. Teratol. 2006, 28, 74–85. [Google Scholar] [CrossRef]
- Yang, D.; Kim, K.H.; Phimister, A.; Bachstetter, A.D.; Ward, T.R.; Stackman, R.W.; Mervis, R.F.; Wisniewski, A.B.; Klein, S.L.; Kodavanti, P.R.; et al. Developmental exposure to polychlorinated biphenyls interferes with experience-dependent dendritic plasticity and ryanodine receptor expression in weanling rats. Environ. Health Perspect. 2009, 117, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayman, G.A.; Yang, D.; Bose, D.D.; Lesiak, A.; Ledoux, V.; Bruun, D.; Pessah, I.N.; Lein, P.J. PCB-95 promotes dendritic growth via ryanodine receptor-dependent mechanisms. Environ. Health Perspect. 2012, 120, 997–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keil, K.P.; Miller, G.W.; Chen, H.; Sethi, S.; Schmuck, M.R.; Dhakal, K.; Kim, J.W.; Lein, P.J. PCB 95 promotes dendritic growth in primary rat hippocampal neurons via mTOR-dependent mechanisms. Arch. Toxicol. 2018, 92, 3163–3173. [Google Scholar] [CrossRef]
- Wayman, G.A.; Bose, D.D.; Yang, D.; Lesiak, A.; Bruun, D.; Impey, S.; Ledoux, V.; Pessah, I.N.; Lein, P.J. PCB-95 modulates the calcium-dependent signaling pathway responsible for activity-dependent dendritic growth. Environ. Health Perspect. 2012, 120, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Kania-Korwel, I.; Ghogha, A.; Chen, H.; Stamou, M.; Bose, D.D.; Pessah, I.N.; Lehmler, H.J.; Lein, P.J. PCB 136 atropselectively alters morphometric and functional parameters of neuronal connectivity in cultured rat hippocampal neurons via ryanodine receptor-dependent mechanisms. Toxicol. Sci. 2014, 138, 379–392. [Google Scholar] [CrossRef]
- Lesiak, A.; Zhu, M.; Chen, H.; Appleyard, S.M.; Impey, S.; Lein, P.J.; Wayman, G.A. The environmental neurotoxicant PCB 95 promotes synaptogenesis via ryanodine receptor-dependent miR132 upregulation. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 717. [Google Scholar] [CrossRef] [PubMed]
- Kimura-Kuroda, J.; Nagata, I.; Kuroda, Y. Disrupting effects of hydroxy-polychlorinated biphenyl (PCB) congeners on neuronal development of cerebellar Purkinje cells: A possible causal factor for developmental brain disorders? Chemosphere 2007, 67, S412–S420. [Google Scholar] [CrossRef]
- Kimura-Kuroda, J.; Nagata, I.; Kuroda, Y. Hydroxylated metabolites of polychlorinated biphenyls inhibit thyroid-hormone-dependent extension of cerebellar Purkinje cell dendrites. Brain Res. Dev. Brain Res. 2005, 154, 259–263. [Google Scholar] [CrossRef]
- Sethi, S.; Keil, K.P.; Lein, P.J. Species and Sex Differences in the Morphogenic Response of Primary Rodent Neurons to 3,3’-Dichlorobiphenyl (PCB 11). Toxics 2017, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Sethi, S.; Keil, K.P.; Lein, P.J. 3,3’-Dichlorobiphenyl (PCB 11) promotes dendritic arborization in primary rat cortical neurons via a CREB-dependent mechanism. Arch. Toxicol. 2018, 92, 3337–3345. [Google Scholar] [CrossRef] [Green Version]
- Kodavanti, P.R.; Tilson, H.A. Neurochemical effects of environmental chemicals: In vitro and in vivo correlations on second messenger pathways. Ann. N. Y. Acad. Sci. 2000, 919, 97–105. [Google Scholar] [CrossRef]
- Yang, J.H.; Kodavanti, P.R. Possible molecular targets of halogenated aromatic hydrocarbons in neuronal cells. Biochem. Biophys. Res. Commun. 2001, 280, 1372–1377. [Google Scholar] [CrossRef]
- Do, Y.; Lee, D.K. Effects of polychlorinated biphenyls on the development of neuronal cells in growth period: Structure-activity relationship. Exp. Neurobiol. 2012, 21, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Inglefield, J.R.; Shafer, T.J. Polychlorinated biphenyl-stimulation of Ca(2+) oscillations in developing neocortical cells: A role for excitatory transmitters and L-type voltage-sensitive Ca(2+) channels. J. Pharmacol. Exp. Ther. 2000, 295, 105–113. [Google Scholar]
- Mundy, W.R.; Shafer, T.J.; Tilson, H.A.; Kodavanti, P.R. Extracellular calcium is required for the polychlorinated biphenyl-induced increase of intracellular free calcium levels in cerebellar granule cell culture. Toxicology 1999, 136, 27–39. [Google Scholar] [CrossRef]
- Inglefield, J.R.; Mundy, W.R.; Shafer, T.J. Inositol 1,4,5-triphosphate receptor-sensitive Ca(2+) release, store-operated Ca(2+) entry, and cAMP responsive element binding protein phosphorylation in developing cortical cells following exposure to polychlorinated biphenyls. J. Pharmacol. Exp. Ther. 2001, 297, 762–773. [Google Scholar] [PubMed]
- Samso, M.; Feng, W.; Pessah, I.N.; Allen, P.D. Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 2009, 7, e85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, E.B.; Feng, W.; Zheng, J.; Dong, Y.; Li, X.; Lehmler, H.J.; Pessah, I.N. An Extended Structure-Activity Relationship of Nondioxin-Like PCBs Evaluates and Supports Modeling Predictions and Identifies Picomolar Potency of PCB 202 Towards Ryanodine Receptors. Toxicol. Sci. 2017, 155, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Zheng, J.; Robin, G.; Dong, Y.; Ichikawa, M.; Inoue, Y.; Mori, T.; Nakano, T.; Pessah, I.N. Enantioselectivity of 2,2’,3,5’,6-Pentachlorobiphenyl (PCB 95) Atropisomers toward Ryanodine Receptors (RyRs) and Their Influences on Hippocampal Neuronal Networks. Environ. Sci. Technol. 2017, 51, 14406–14416. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, E.B.; Pessah, I.N. Structure-activity relationship of non-coplanar polychlorinated biphenyls toward skeletal muscle ryanodine receptors in rainbow trout (Oncorhynchus mykiss). Aquat. Toxicol. 2013, 140–141, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, E.B.; Stegeman, J.J.; Goldstone, J.V.; Nacci, D.E.; Champlin, D.; Jayaraman, S.; Connon, R.E.; Pessah, I.N. Expression and function of ryanodine receptor related pathways in PCB tolerant Atlantic killifish (Fundulus heteroclitus) from New Bedford Harbor, MA, USA. Aquat. Toxicol. 2015, 159, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Holland, E.B.; Goldstone, J.V.; Pessah, I.N.; Whitehead, A.; Reid, N.M.; Karchner, S.I.; Hahn, M.E.; Nacci, D.E.; Clark, B.W.; Stegeman, J.J. Ryanodine receptor and FK506 binding protein 1 in the Atlantic killifish (Fundulus heteroclitus): A phylogenetic and population-based comparison. Aquat. Toxicol. 2017, 192, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Niknam, Y.; Feng, W.; Cherednichenko, G.; Dong, Y.; Joshi, S.N.; Vyas, S.M.; Lehmler, H.J.; Pessah, I.N. Structure-activity relationship of selected meta- and para-hydroxylated non-dioxin like polychlorinated biphenyls: From single RyR1 channels to muscle dysfunction. Toxicol. Sci. 2013, 136, 500–513. [Google Scholar] [CrossRef]
- Pessah, I.N.; Hansen, L.G.; Albertson, T.E.; Garner, C.E.; Ta, T.A.; Do, Z.; Kim, K.H.; Wong, P.W. Structure-activity relationship for noncoplanar polychlorinated biphenyl congeners toward the ryanodine receptor-Ca2+ channel complex type 1 (RyR1). Chem. Res. Toxicol. 2006, 19, 92–101. [Google Scholar] [CrossRef]
- Wong, P.W.; Brackney, W.R.; Pessah, I.N. Ortho-substituted polychlorinated biphenyls alter microsomal calcium transport by direct interaction with ryanodine receptors of mammalian brain. J. Biol. Chem. 1997, 272, 15145–15153. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.W.; Pessah, I.N. Ortho-substituted polychlorinated biphenyls alter calcium regulation by a ryanodine receptor-mediated mechanism: Structural specificity toward skeletal- and cardiac-type microsomal calcium release channels. Mol. Pharmacol. 1996, 49, 740–751. [Google Scholar]
- Schratt, G.M.; Nigh, E.A.; Chen, W.G.; Hu, L.; Greenberg, M.E. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J. Neurosci. 2004, 24, 7366–7377. [Google Scholar] [CrossRef]
- Tsokas, P.; Grace, E.A.; Chan, P.; Ma, T.; Sealfon, S.C.; Iyengar, R.; Landau, E.M.; Blitzer, R.D. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J. Neurosci. 2005, 25, 5833–5843. [Google Scholar] [CrossRef]
- Wayman, G.A.; Impey, S.; Marks, D.; Saneyoshi, T.; Grant, W.F.; Derkach, V.; Soderling, T.R. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 2006, 50, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Kenet, T.; Froemke, R.C.; Schreiner, C.E.; Pessah, I.N.; Merzenich, M.M. Perinatal exposure to a noncoplanar polychlorinated biphenyl alters tonotopy, receptive fields, and plasticity in rat primary auditory cortex. Proc. Natl. Acad. Sci. USA 2007, 104, 7646–7651. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, L.; Llansola, M.; Felipo, V.; Campagna, R.; Airoldi, L.; De Paola, M.; Fanelli, R.; Mariani, A.; Mazzoletti, M.; Pastorelli, R. Insight into the neuroproteomics effects of the food-contaminant non-dioxin like polychlorinated biphenyls. J. Proteom. 2012, 75, 2417–2430. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Irwin, S.A.; Patel, B.; Idupulapati, M.; Harris, J.B.; Crisostomo, R.A.; Larsen, B.P.; Kooy, F.; Willems, P.J.; Cras, P.; Kozlowski, P.B.; et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am. J. Med. Genet. 2001, 98, 161–167. [Google Scholar] [CrossRef]
- Cheslack-Postava, K.; Rantakokko, P.V.; Hinkka-Yli-Salomaki, S.; Surcel, H.M.; McKeague, I.W.; Kiviranta, H.A.; Sourander, A.; Brown, A.S. Maternal serum persistent organic pollutants in the Finnish Prenatal Study of Autism: A pilot study. Neurotoxicol. Teratol. 2013, 38, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Lu, Z.; Li, G.; Piechowicz, M.; Anderson, M.; Uddin, Y.; Wu, J.; Qiu, S. The autism-associated MET receptor tyrosine kinase engages early neuronal growth mechanism and controls glutamatergic circuits development in the forebrain. Mol. Psychiatry 2016, 21, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Volk, H.E.; Kerin, T.; Lurmann, F.; Hertz-Picciotto, I.; McConnell, R.; Campbell, D.B. Autism spectrum disorder: Interaction of air pollution with the MET receptor tyrosine kinase gene. Epidemiology 2014, 25, 44–47. [Google Scholar] [CrossRef] [Green Version]
- Dunaway, K.W.; Islam, M.S.; Coulson, R.L.; Lopez, S.J.; Vogel Ciernia, A.; Chu, R.G.; Yasui, D.H.; Pessah, I.N.; Lott, P.; Mordaunt, C.; et al. Cumulative Impact of Polychlorinated Biphenyl and Large Chromosomal Duplications on DNA Methylation, Chromatin, and Expression of Autism Candidate Genes. Cell Rep. 2016, 17, 3035–3048. [Google Scholar] [CrossRef]
- Krey, J.F.; Dolmetsch, R.E. Molecular mechanisms of autism: A possible role for Ca2+ signaling. Curr. Opin. Neurobiol. 2007, 17, 112–119. [Google Scholar] [CrossRef]
- Marcantoni, A.; Calorio, C.; Hidisoglu, E.; Chiantia, G.; Carbone, E. Cav1.2 channelopathies causing autism: New hallmarks on Timothy syndrome. Pflug. Arch. 2020, 472, 775–789. [Google Scholar] [CrossRef]
- Nguyen, R.L.; Medvedeva, Y.V.; Ayyagari, T.E.; Schmunk, G.; Gargus, J.J. Intracellular calcium dysregulation in autism spectrum disorder: An analysis of converging organelle signaling pathways. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1718–1732. [Google Scholar] [CrossRef]
- Robin, G.; Lopez, J.R.; Espinal, G.M.; Hulsizer, S.; Hagerman, P.J.; Pessah, I.N. Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2017, 26, 2649–2666. [Google Scholar] [CrossRef] [Green Version]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Pasca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Pasca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Leehey, M.A.; Hagerman, P.J. Fragile X-associated tremor/ataxia syndrome. Handb. Clin. Neurol. 2012, 103, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Krueger, D.D.; Bear, M.F. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med. 2011, 62, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Hulsizer, S.; Tassone, F.; Tang, H.T.; Hagerman, R.J.; Rogawski, M.A.; Hagerman, P.J.; Pessah, I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: Amelioration with allopregnanolone. Hum. Mol. Genet. 2012, 21, 2923–2935. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Hagerman, R.J.; Willemsen, R.; Pessah, I.N. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 2010, 19, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.; Hagerman, P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013, 12, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Eagleson, K.L.; Xie, Z.; Levitt, P. The Pleiotropic MET Receptor Network: Circuit Development and the Neural-Medical Interface of Autism. Biol. Psychiatry 2017, 81, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef]
- Kalkman, H.O. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol. Autism 2012, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinson, H.; Nakatani, J.; Babineau, B.A.; Birnbaum, R.Y.; Ellegood, J.; Bershteyn, M.; McEvilly, R.J.; Long, J.M.; Willert, K.; Klein, O.D.; et al. Prenatal beta-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol. Psychiatry 2016, 21, 1417–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, Q.; Wang, A.; Hamzah, H.; Waldman, A.; Jiang, K.; Dong, Q.; Li, R.; Kim, J.; Turner, D.; Chang, Q. CREB Signaling Is Involved in Rett Syndrome Pathogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 3671–3685. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Doering, L.C. Reversing autism by targeting downstream mTOR signaling. Front. Cell Neurosci. 2013, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Li, R.J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. Elife 2016, 5. [Google Scholar] [CrossRef]
- Todd, P.K.; Mack, K.J. Phosphorylation, CREB, and Mental Retardation. Pediatric Res. 2001, 50, 672. [Google Scholar] [CrossRef]
- Ngounou Wetie, A.G.; Wormwood, K.L.; Charette, L.; Ryan, J.P.; Woods, A.G.; Darie, C.C. Comparative two-dimensional polyacrylamide gel electrophoresis of the salivary proteome of children with autism spectrum disorder. J. Cell Mol. Med. 2015, 19, 2664–2678. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Kasper, L.H.; Bedford, D.C.; Lerach, S.; Teubner, B.J.; Brindle, P.K. Mutation of the CH1 Domain in the Histone Acetyltransferase CREBBP Results in Autism-Relevant Behaviors in Mice. PLoS ONE 2016, 11, e0146366. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elneel, K.; Liu, T.; Gazzaniga, F.S.; Nishimura, Y.; Wall, D.P.; Geschwind, D.H.; Lao, K.; Kosik, K.S. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 2008, 9, 153–161. [Google Scholar] [CrossRef]
- Sarachana, T.; Zhou, R.; Chen, G.; Manji, H.K.; Hu, V.W. Investigation of post-transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Med. 2010, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Talebizadeh, Z.; Butler, M.G.; Theodoro, M.F. Feasibility and relevance of examining lymphoblastoid cell lines to study role of microRNAs in autism. Autism Res. Off. J. Int. Soc. Autism Res. 2008, 1, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Bocchi, R.; Egervari, K.; Carol-Perdiguer, L.; Viale, B.; Quairiaux, C.; De Roo, M.; Boitard, M.; Oskouie, S.; Salmon, P.; Kiss, J.Z. Perturbed Wnt signaling leads to neuronal migration delay, altered interhemispheric connections and impaired social behavior. Nat. Commun. 2017, 8, 1158. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.K.; Bourgeron, T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat. Neurosci. 2006, 9, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387, 167–178. [Google Scholar] [CrossRef]
- Hansen, K.F.; Karelina, K.; Sakamoto, K.; Wayman, G.A.; Impey, S.; Obrietan, K. miRNA-132: A dynamic regulator of cognitive capacity. Brain Struct. Funct. 2013, 218, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Dardiotis, E.; Aloizou, A.M.; Siokas, V.; Tsouris, Z.; Rikos, D.; Marogianni, C.; Aschner, M.; Kovatsi, L.; Bogdanos, D.P.; Tsatsakis, A. Paraoxonase-1 genetic polymorphisms in organophosphate metabolism. Toxicology 2019, 411, 24–31. [Google Scholar] [CrossRef]
- Mordaunt, C.E.; Park, B.Y.; Bakulski, K.M.; Feinberg, J.I.; Croen, L.A.; Ladd-Acosta, C.; Newschaffer, C.J.; Volk, H.E.; Ozonoff, S.; Hertz-Picciotto, I.; et al. A meta-analysis of two high-risk prospective cohort studies reveals autism-specific transcriptional changes to chromatin, autoimmune, and environmental response genes in umbilical cord blood. Mol. Autism 2019, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Warner, N.A.; Martin, J.W.; Wong, C.S. Chiral polychlorinated biphenyls are biotransformed enantioselectively by mammalian cytochrome P-450 isozymes to form hydroxylated metabolites. Environ. Sci. Technol. 2009, 43, 114–121. [Google Scholar] [CrossRef]
- Dreiem, A.; Rykken, S.; Lehmler, H.J.; Robertson, L.W.; Fonnum, F. Hydroxylated polychlorinated biphenyls increase reactive oxygen species formation and induce cell death in cultured cerebellar granule cells. Toxicol. Appl. Pharmacol. 2009, 240, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tan, Y.; Song, E.; Song, Y. A Critical Review of Polychlorinated Biphenyls Metabolism, Metabolites, and Their Correlation with Oxidative Stress. Chem. Res. Toxicol. 2020, 33, 2022–2042. [Google Scholar] [CrossRef]
- Vichi, S.; Medda, E.; Ingelido, A.M.; Ferro, A.; Resta, S.; Porpora, M.G.; Abballe, A.; Nistico, L.; De Felip, E.; Gemma, S.; et al. Glutathione transferase polymorphisms and risk of endometriosis associated with polychlorinated biphenyls exposure in Italian women: A gene-environment interaction. Fertil. Steril. 2012, 97, 1143–1151.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roya, R.; Baludu, G.S.; Reddy, B.S. Possible aggravating impact of gene polymorphism in women with endometriosis. Indian J. Med. Res. 2009, 129, 395–400. [Google Scholar] [PubMed]
- Everson, J.L.; Sun, M.R.; Fink, D.M.; Heyne, G.W.; Melberg, C.G.; Nelson, K.F.; Doroodchi, P.; Colopy, L.J.; Ulschmid, C.M.; Martin, A.A.; et al. Developmental Toxicity Assessment of Piperonyl Butoxide Exposure Targeting Sonic Hedgehog Signaling and Forebrain and Face Morphogenesis in the Mouse: An in Vitro and in Vivo Study. Environ. Health Perspect. 2019, 127, 107006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Jarvik, G.P.; Browning, B.L.; Rajagopalan, R.; Gordon, A.S.; Rieder, M.J.; Robertson, P.D.; Nickerson, D.A.; Fisher, N.A.; Hopkins, P.M. Exome sequencing reveals novel rare variants in the ryanodine receptor and calcium channel genes in malignant hyperthermia families. Anesthesiology 2013, 119, 1054–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, T.A.; Pessah, I.N. Ryanodine receptor type 1 (RyR1) possessing malignant hyperthermia mutation R615C exhibits heightened sensitivity to dysregulation by non-coplanar 2,2’,3,5’,6-pentachlorobiphenyl (PCB 95). Neurotoxicology 2007, 28, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Cantor, R.M. Allowing for sex differences increases power in a GWAS of multiplex Autism families. Mol. Psychiatry 2012, 17, 215–222. [Google Scholar] [CrossRef]
- Barrientos, G.C.; Feng, W.; Truong, K.; Matthaei, K.I.; Yang, T.; Allen, P.D.; Lopez, J.R.; Pessah, I.N. Gene dose influences cellular and calcium channel dysregulation in heterozygous and homozygous T4826I-RYR1 malignant hyperthermia-susceptible muscle. J. Biol. Chem. 2012, 287, 2863–2876. [Google Scholar] [CrossRef] [Green Version]
- Yuen, B.; Boncompagni, S.; Feng, W.; Yang, T.; Lopez, J.R.; Matthaei, K.I.; Goth, S.R.; Protasi, F.; Franzini-Armstrong, C.; Allen, P.D.; et al. Mice expressing T4826I-RYR1 are viable but exhibit sex- and genotype-dependent susceptibility to malignant hyperthermia and muscle damage. FASEB J. 2012, 26, 1311–1322. [Google Scholar] [CrossRef] [Green Version]
- Keil, K.P.; Sethi, S.; Wilson, M.D.; Silverman, J.L.; Pessah, I.N.; Lein, P.J. Genetic mutations in Ca(2+) signaling alter dendrite morphology and social approach in juvenile mice. Genes Brain Behav. 2019, 18, e12526. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Schmidt, R.J.; Walker, C.K.; Bennett, D.H.; Oliver, M.; Shedd-Wise, K.M.; LaSalle, J.M.; Giulivi, C.; Puschner, B.; Thomas, J.; et al. A Prospective Study of Environmental Exposures and Early Biomarkers in Autism Spectrum Disorder: Design, Protocols, and Preliminary Data from the MARBLES Study. Environ. Health Perspect. 2018, 126, 117004. [Google Scholar] [CrossRef] [Green Version]
- Rude, K.M.; Keogh, C.E.; Gareau, M.G. The role of the gut microbiome in mediating neurotoxic outcomes to PCB exposure. Neurotoxicology 2019, 75, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Julu, P.O.; Brimacombe, M.; Connor, S.; Daniels, M.L. Reduced cardiac parasympathetic activity in children with autism. Brain Dev. 2005, 27, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Toichi, M.; Kamio, Y. Paradoxical autonomic response to mental tasks in autism. J. Autism Dev. Disord. 2003, 33, 417–426. [Google Scholar] [CrossRef]
- Lory, C.; Kadlaskar, G.; McNally Keehn, R.; Francis, A.L.; Keehn, B. Brief Report: Reduced Heart Rate Variability in Children with Autism Spectrum Disorder. J. Autism Dev. Disord 2020. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Huang, Y.C.; Huang, W.L. Heart rate variability in individuals with autism spectrum disorders: A meta-analysis. Neurosci. Biobehav. Rev. 2020, 118, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Hirstein, W.; Iversen, P.; Ramachandran, V.S. Autonomic responses of autistic children to people and objects. Proc. Biol. Sci. 2001, 268, 1883–1888. [Google Scholar] [CrossRef]
- Haigh, S.M.; Endevelt-Shapira, Y.; Behrmann, M. Trial-to-Trial Variability in Electrodermal Activity to Odor in Autism. Autism Res. 2020. [Google Scholar] [CrossRef]
- Schoen, S.A.; Miller, L.J.; Brett-Green, B.A.; Nielsen, D.M. Physiological and behavioral differences in sensory processing: A comparison of children with autism spectrum disorder and sensory modulation disorder. Front. Integr. Neurosci. 2009, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Zhu, J.; Yang, T.; Wang, S.; Liu, H.; Wu, Q.; Zhang, X.; Chen, J.; Li, T. Vitamin A deficiency exacerbates autism-like behaviors and abnormalities of the enteric nervous system in a valproic acid-induced rat model of autism. Neurotoxicology 2020, 79, 184–190. [Google Scholar] [CrossRef]
- Chen, R.C.; Tang, S.Y.; Miyata, H.; Kashimoto, T.; Chang, Y.C.; Chang, K.J.; Tung, T.C. Polychlorinated biphenyl poisoning: Correlation of sensory and motor nerve conduction, neurologic symptoms, and blood levels of polychlorinated biphenyls, quaterphenyls, and dibenzofurans. Environ. Res. 1985, 37, 340–348. [Google Scholar] [CrossRef]
- Boegner, F.; Franke, D.; Altenkirch, H.; Stoltenburg, G.; Wagner, M. PCBs have a predominantly neurotoxic effect on dissociated cultures of the nervous system. Neurotoxicology 1994, 15, 593–596. [Google Scholar] [PubMed]
- Raffetti, E.; Donato, F.; De Palma, G.; Leonardi, L.; Sileo, C.; Magoni, M. Polychlorinated biphenyls (PCBs) and risk of hypertension: A population-based cohort study in a North Italian highly polluted area. Sci. Total Environ. 2020, 714, 136660. [Google Scholar] [CrossRef]
- Pavuk, M.; Serio, T.C.; Cusack, C.; Cave, M.; Rosenbaum, P.F.; Birnbaum, L.S. Hypertension in Relation to Dioxins and Polychlorinated Biphenyls from the Anniston Community Health Survey Follow-Up. Environ. Health Perspect. 2019, 127, 127007. [Google Scholar] [CrossRef]
- Saxena, T.; Ali, A.O.; Saxena, M. Pathophysiology of essential hypertension: An update. Expert Rev. Cardiovasc. Ther. 2018, 16, 879–887. [Google Scholar] [CrossRef]
- Ivanov, A.; Purves, D. Ongoing electrical activity of superior cervical ganglion cells in mammals of different size. J. Comp. Neurol. 1989, 284, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Gonsiorek, E.A.; Barnhart, C.; Davare, M.A.; Engebose, A.J.; Lauridsen, H.; Bruun, D.; Lesiak, A.; Wayman, G.; Bucelli, R.; et al. Statins decrease dendritic arborization in rat sympathetic neurons by blocking RhoA activation. J. Neurochem. 2009, 108, 1057–1071. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Terada, M.; Shimizu, D.; Fujiwara, T.; Tabei, R. Morphometric study of the superior cervical and stellate ganglia of spontaneously hypertensive rats during the prehypertensive stage. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1990, 58, 371–376. [Google Scholar] [CrossRef]
- Muller, P.A.; Schneeberger, M.; Matheis, F.; Wang, P.; Kerner, Z.; Ilanges, A.; Pellegrino, K.; Del Marmol, J.; Castro, T.B.R.; Furuichi, M.; et al. Microbiota modulate sympathetic neurons via a gut-brain circuit. Nature 2020. [Google Scholar] [CrossRef]
- Lein, P.J.; Fryer, A.D. Autonomic and enteric neurons: Cell culture. In Reference Module in Neuroscience and Biobehavioral Psychology; Stein, J., Ed.; Elsevier Ltd.: Oxford, UK, 2019. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618.e17. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.R.; Minuto, C.; Cryan, J.F.; Clarke, G.; Dinan, T.G. Cross Talk: The Microbiota and Neurodevelopmental Disorders. Front. Neurosci. 2017, 11, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Y.; Lin, Y.; Lu, Y.; Huang, Q.; Ye, G.; Dong, S. Gut microbiota dysbiosis correlates with a low-dose PCB126-induced dyslipidemia and non-alcoholic fatty liver disease. Sci. Total Environ. 2019, 653, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Wang, H.; Lin, Y.; Lu, Y.; Huang, Q.; Ye, G.; Dong, S. Gut microbiota characterization and lipid metabolism disorder found in PCB77-treated female mice. Toxicology 2019, 420, 11–20. [Google Scholar] [CrossRef]
- Laue, H.E.; Brennan, K.J.M.; Gillet, V.; Abdelouahab, N.; Coull, B.A.; Weisskopf, M.G.; Burris, H.H.; Zhang, W.; Takser, L.; Baccarelli, A.A. Associations of prenatal exposure to polybrominated diphenyl ethers and polychlorinated biphenyls with long-term gut microbiome structure: A pilot study. Environ. Epidemiol. 2019, 3. [Google Scholar] [CrossRef]
- Lim, J.J.; Li, X.; Lehmler, H.J.; Wang, D.; Gu, H.; Cui, J.Y. Gut Microbiome Critically Impacts PCB-induced Changes in Metabolic Fingerprints and the Hepatic Transcriptome in Mice. Toxicol. Sci. 2020. [Google Scholar] [CrossRef]
- Rude, K.M.; Pusceddu, M.M.; Keogh, C.E.; Sladek, J.A.; Rabasa, G.; Miller, E.N.; Sethi, S.; Keil, K.P.; Pessah, I.N.; Lein, P.J.; et al. Developmental exposure to polychlorinated biphenyls (PCBs) in the maternal diet causes host-microbe defects in weanling offspring mice. Environ. Pollut. 2019, 253, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlang, B.; Barney, J.; Thompson, B.; Wang, C.; Hamad, O.M.; Hoffman, J.B.; Petriello, M.C.; Morris, A.J.; Hennig, B. Editor’s Highlight: PCB126 Exposure Increases Risk for Peripheral Vascular Diseases in a Liver Injury Mouse Model. Toxicol. Sci. 2017, 160, 256–267. [Google Scholar] [CrossRef]
- Wahlang, B.; Jin, J.; Hardesty, J.E.; Head, K.Z.; Shi, H.; Falkner, K.C.; Prough, R.A.; Klinge, C.M.; Cave, M.C. Identifying sex differences arising from polychlorinated biphenyl exposures in toxicant-associated liver disease. Food Chem. Toxicol. 2019, 129, 64–76. [Google Scholar] [CrossRef]
- Hennig, B.; Reiterer, G.; Toborek, M.; Matveev, S.V.; Daugherty, A.; Smart, E.; Robertson, L.W. Dietary fat interacts with PCBs to induce changes in lipid metabolism in mice deficient in low-density lipoprotein receptor. Environ. Health Perspect. 2005, 113, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Petriello, M.C.; Brandon, J.A.; Hoffman, J.; Wang, C.; Tripathi, H.; Abdel-Latif, A.; Ye, X.; Li, X.; Yang, L.; Lee, E.; et al. Dioxin-like PCB 126 Increases Systemic Inflammation and Accelerates Atherosclerosis in Lean LDL Receptor-Deficient Mice. Toxicol. Sci. 2018, 162, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Petriello, M.C.; Zhu, B.; Hennig, B. PCB 126 induces monocyte/macrophage polarization and inflammation through AhR and NF-kappaB pathways. Toxicol. Appl. Pharmacol. 2019, 367, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Gubbiotti, M.; Balboni, G.; Bini, V.; Elisei, S.; Bedetti, C.; Marchiafava, M.; Giannantoni, A. Bladder and bowel dysfunction, adaptive behaviour and psychiatric profiles in adults affected by autism spectrum disorders. Neurourol. Urodyn. 2019, 38, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Gubbiotti, M.; Elisei, S.; Bedetti, C.; Marchiafava, M.; Giannantoni, A. Urinary and bowel disfunction in autism spectrum disorder: A prospective, observational study. Psychiatr. Danub. 2019, 31, 475–478. [Google Scholar]
- Kostiukow, A.; Poniewierski, P.; Daroszewski, P.; Samborski, W. Gastrointestinal disorders in children with autism spectrum disorder. Polski Merkuriusz Lekarski 2020, 48, 69–72. [Google Scholar] [PubMed]
- Von Gontard, A.; Pirrung, M.; Niemczyk, J.; Equit, M. Incontinence in children with autism spectrum disorder. J. Pediatr. Urol. 2015, 11, 264.e1–264.e7. [Google Scholar] [CrossRef]
- Von Gontard, A.; Baeyens, D.; Van Hoecke, E.; Warzak, W.J.; Bachmann, C. Psychological and psychiatric issues in urinary and fecal incontinence. J. Urol. 2011, 185, 1432–1436. [Google Scholar] [CrossRef]
- Alabaf, S.; Gillberg, C.; Lundstrom, S.; Lichtenstein, P.; Kerekes, N.; Rastam, M.; Anckarsater, H. Physical health in children with neurodevelopmental disorders. J. Autism Dev. Disord. 2019, 49, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Von Gontard, A.; Equit, M. Comorbidity of ADHD and incontinence in children. Eur. Child. Adolesc. Psychiatry 2015, 24, 127–140. [Google Scholar] [CrossRef]
- Elia, J.; Takeda, T.; Deberardinis, R.; Burke, J.; Accardo, J.; Ambrosini, P.J.; Blum, N.J.; Brown, L.W.; Lantieri, F.; Berrettini, W.; et al. Nocturnal enuresis: A suggestive endophenotype marker for a subgroup of inattentive attention-deficit/hyperactivity disorder. J. Pediatric 2009, 155, 239–244.e5. [Google Scholar] [CrossRef]
- Taylor, J.A.; Jones, M.B.; Besch-Williford, C.L.; Berendzen, A.F.; Ricke, W.A.; vom Saal, F.S. Interactive Effects of Perinatal BPA or DES and Adult Testosterone and Estradiol Exposure on Adult Urethral Obstruction and Bladder, Kidney, and Prostate Pathology in Male Mice. Int. J. Mol. Sci. 2020, 21, 3902. [Google Scholar] [CrossRef]
- Turco, A.E.; Thomas, S.; Crawford, L.K.; Tang, W.; Peterson, R.E.; Li, L.; Ricke, W.A.; Vezina, C.M. In utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure exacerbates urinary dysfunction in hormone-treated C57BL/6J mice through a non-malignant mechanism involving proteomic changes in the prostate that differ from those elicited by testosterone and estradiol. Am. J. Clin. Exp. Urol. 2020, 8, 59–72. [Google Scholar] [PubMed]
- Ricke, W.A.; Lee, C.W.; Clapper, T.R.; Schneider, A.J.; Moore, R.W.; Keil, K.P.; Abler, L.L.; Wynder, J.L.; Lopez Alvarado, A.; Beaubrun, I.; et al. In Utero and Lactational TCDD Exposure Increases Susceptibility to Lower Urinary Tract Dysfunction in Adulthood. Toxicol. Sci 2016, 150, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, T.M.; Nguyen, J.L.; Leverson, G.E.; Taylor, J.A.; Vom Saal, F.S.; Wood, R.W.; Ricke, W.A. Endocrine disruptor bisphenol A is implicated in urinary voiding dysfunction in male mice. Am. J. Physiol. Ren. Physiol. 2018, 315, F1208–F1216. [Google Scholar] [CrossRef]
- Fritz, N.; Morel, J.L.; Jeyakumar, L.H.; Fleischer, S.; Allen, P.D.; Mironneau, J.; Macrez, N. RyR1-specific requirement for depolarization-induced Ca2+ sparks in urinary bladder smooth muscle. J. Cell Sci. 2007, 120, 3784–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteoli, G.; Boeckxstaens, G.E. The vagal innervation of the gut and immune homeostasis. Gut 2013, 62, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Roy, H.A.; Green, A.L. The Central Autonomic Network and Regulation of Bladder Function. Front. Neurosci. 2019, 13, 535. [Google Scholar] [CrossRef]
- Birder, L.A.; Kullmann, A.F.; Chapple, C.R. The aging bladder insights from animal models. Asian J. Urol. 2018, 5, 135–140. [Google Scholar] [CrossRef]
- Birder, L.A.; de Groat, W.C. Mechanisms of disease: Involvement of the urothelium in bladder dysfunction. Nat. Clin. Pr. Urol. 2007, 4, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.H.; Hyun, M.; Taranda, J.; Huang, K.W.; Todd, E.; Feng, D.; Atwater, E.; Croney, D.; Zeidel, M.L.; Osten, P.; et al. Central Control Circuit for Context-Dependent Micturition. Cell 2016, 167, 73–86.e12. [Google Scholar] [CrossRef] [Green Version]
- Tykocki, N.R.; Heppner, T.J.; Erickson, C.S.; van Batavia, J.; Vizzard, M.A.; Nelson, M.T.; Mingin, G.C. Development of stress-induced bladder insufficiency requires functional TRPV1 channels. Am. J. Physiol. Ren. Physiol. 2018, 315, F1583–F1591. [Google Scholar] [CrossRef]
- Daly, D.; Rong, W.; Chess-Williams, R.; Chapple, C.; Grundy, D. Bladder afferent sensitivity in wild-type and TRPV1 knockout mice. J. Physiol. 2007, 583, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Keil, K.P.; Abler, L.L.; Altmann, H.M.; Bushman, W.; Marker, P.C.; Li, L.; Ricke, W.A.; Bjorling, D.E.; Vezina, C.M. Influence of animal husbandry practices on void spot assay outcomes in C57BL/6J male mice. Neurourol. Urodyn. 2016, 35, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Guzman, E.C.; Nimunkar, A.; Keil, K.P.; Vezina, C.M.; Ricke, W.A.; Macoska, J.; Bjorling, D.E. Void sorcerer: An open source, open access framework for mouse uroflowmetry. Am. J. Clin. Exp. Urol. 2019, 7, 170–177. [Google Scholar]
- Wegner, K.A.; Abler, L.L.; Oakes, S.R.; Mehta, G.S.; Ritter, K.E.; Hill, W.G.; Zwaans, B.M.; Lamb, L.E.; Wang, Z.; Bjorling, D.E.; et al. Void spot assay procedural optimization and software for rapid and objective quantification of rodent voiding function, including overlapping urine spots. Am. J. Physiol. Ren. Physiol. 2018, 315, F1067–F1080. [Google Scholar] [CrossRef]
- Grodzki, A.C.; Ghogha, A.; Mangini, L.; Fryer, A.D.; Lein, P.J. IFNgamma Increases M2 Muscarinic Receptor Expression in Cultured Sympathetic Neurons. Curr. Neurobiol. 2011, 2, 23–29. [Google Scholar]
- Yang, D.; Howard, A.; Bruun, D.; Ajua-Alemanj, M.; Pickart, C.; Lein, P.J. Chlorpyrifos and chlorpyrifos-oxon inhibit axonal growth by interfering with the morphogenic activity of acetylcholinesterase. Toxicol. Appl. Pharmacol. 2008, 228, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Kania-Korwel, I.; Lehmler, H.J.; Wong, C.S. Stereoselective formation of mono- and dihydroxylated polychlorinated biphenyls by rat cytochrome P450 2B1. Environ. Sci. Technol. 2013, 47, 12184–12192. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Wong, C.S. Factors affecting phase I stereoselective biotransformation of chiral polychlorinated biphenyls by rat cytochrome P-450 2B1 isozyme. Environ. Sci. Technol. 2011, 45, 8298–8305. [Google Scholar] [CrossRef]
- Wu, X.; Duffel, M.; Lehmler, H.J. Oxidation of polychlorinated biphenyls by liver tissue slices from phenobarbital-pretreated mice is congener-specific and atropselective. Chem. Res. Toxicol. 2013, 26, 1642–1651. [Google Scholar] [CrossRef]
- Wu, X.; Kania-Korwel, I.; Chen, H.; Stamou, M.; Dammanahalli, K.J.; Duffel, M.; Lein, P.J.; Lehmler, H.J. Metabolism of 2,2’,3,3’,6,6’-hexachlorobiphenyl (PCB 136) atropisomers in tissue slices from phenobarbital or dexamethasone-induced rats is sex-dependent. Xenobiotica 2013, 43, 933–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Pramanik, A.; Duffel, M.W.; Hrycay, E.G.; Bandiera, S.M.; Lehmler, H.J.; Kania-Korwel, I. 2,2’,3,3’,6,6’-Hexachlorobiphenyl (PCB 136) is enantioselectively oxidized to hydroxylated metabolites by rat liver microsomes. Chem. Res. Toxicol. 2011, 24, 2249–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.Y.; Gu, J.; Su, T.; Cui, H.; Zhang, X.; D’Agostino, J.; Zhuo, X.; Yang, W.; Swiatek, P.J.; Ding, X. Generation and characterization of a transgenic mouse model with hepatic expression of human CYP2A6. Biochem. Biophys. Res. Commun. 2005, 338, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Carratt, S.; Hartog, M.; Kovalchik, N.; Jia, K.; Wang, Y.; Zhang, Q.Y.; Edwards, P.; Winkle, L.V.; Ding, X. Human CYP2A13 and CYP2F1 Mediate Naphthalene Toxicity in the Lung and Nasal Mucosa of CYP2A13/2F1-Humanized Mice. Environ. Health Perspect. 2017, 125, 067004. [Google Scholar] [CrossRef]
- Li, L.; Megaraj, V.; Wei, Y.; Ding, X. Identification of cytochrome P450 enzymes critical for lung tumorigenesis by the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK): Insights from a novel Cyp2abfgs-null mouse. Carcinogenesis 2014, 35, 2584–2591. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Li, L.; Zhou, X.; Zhang, Q.Y.; Dunbar, A.; Liu, F.; Kluetzman, K.; Yang, W.; Ding, X. Generation and characterization of a novel Cyp2a(4/5)bgs-null mouse model. Drug Metab. Dispos. 2013, 41, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Wu, H.; Li, L.; Liu, Z.; Zhou, X.; Zhang, Q.Y.; Weng, Y.; D’Agostino, J.; Ling, G.; Zhang, X.; et al. Generation and characterization of a CYP2A13/2B6/2F1-transgenic mouse model. Drug Metab. Dispos. 2012, 40, 1144–1150. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, L.; Wu, H.; Hu, J.; Ma, J.; Zhang, Q.Y.; Ding, X. Characterization of CYP2B6 in a CYP2B6-humanized mouse model: Inducibility in the liver by phenobarbital and dexamethasone and role in nicotine metabolism in vivo. Drug Metab. Dispos. 2015, 43, 208–216. [Google Scholar] [CrossRef] [Green Version]
- McGraw, J.E., Sr.; Waller, D.P. Specific human CYP 450 isoform metabolism of a pentachlorobiphenyl (PCB-IUPAC# 101). Biochem. Biophys. Res. Commun. 2006, 344, 129–133. [Google Scholar] [CrossRef]
- Shimada, T.; Kakimoto, K.; Takenaka, S.; Koga, N.; Uehara, S.; Murayama, N.; Yamazaki, H.; Kim, D.; Guengerich, F.P.; Komori, M. Roles of Human CYP2A6 and Monkey CYP2A24 and 2A26 Cytochrome P450 Enzymes in the Oxidation of 2,5,2’,5’-Tetrachlorobiphenyl. Drug Metab. Dispos. 2016, 44, 1899–1909. [Google Scholar] [CrossRef] [Green Version]
- Ariyoshi, N.; Oguri, K.; Koga, N.; Yoshimura, H.; Funae, Y. Metabolism of highly persistent PCB congener, 2,4,5,2’,4’,5’-hexachlorobiphenyl, by human CYP2B6. Biochem. Biophys. Res. Commun. 1995, 212, 455–460. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Factor | ASD-Relevant Genetic Risk Factors | Further Reading |
|---|---|---|
| Genetic variants or changes in expression of genes regulating calcium signaling or gene products regulated by calcium |
| |
| Genetic variants or changes in expression of genes regulating dendritic arborization, axonal growth, or synapses |
| |
| Genetic polymorphisms that alter metabolism or response to PCBs |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panesar, H.K.; Kennedy, C.L.; Keil Stietz, K.P.; Lein, P.J. Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder? Toxics 2020, 8, 70. https://doi.org/10.3390/toxics8030070
Panesar HK, Kennedy CL, Keil Stietz KP, Lein PJ. Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder? Toxics. 2020; 8(3):70. https://doi.org/10.3390/toxics8030070
Chicago/Turabian StylePanesar, Harmanpreet Kaur, Conner L. Kennedy, Kimberly P. Keil Stietz, and Pamela J. Lein. 2020. "Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder?" Toxics 8, no. 3: 70. https://doi.org/10.3390/toxics8030070
APA StylePanesar, H. K., Kennedy, C. L., Keil Stietz, K. P., & Lein, P. J. (2020). Polychlorinated Biphenyls (PCBs): Risk Factors for Autism Spectrum Disorder? Toxics, 8(3), 70. https://doi.org/10.3390/toxics8030070