
Adrenergic and Glucocorticoid Receptors in the Pulmonary Health Effects of Air Pollution

Abstract
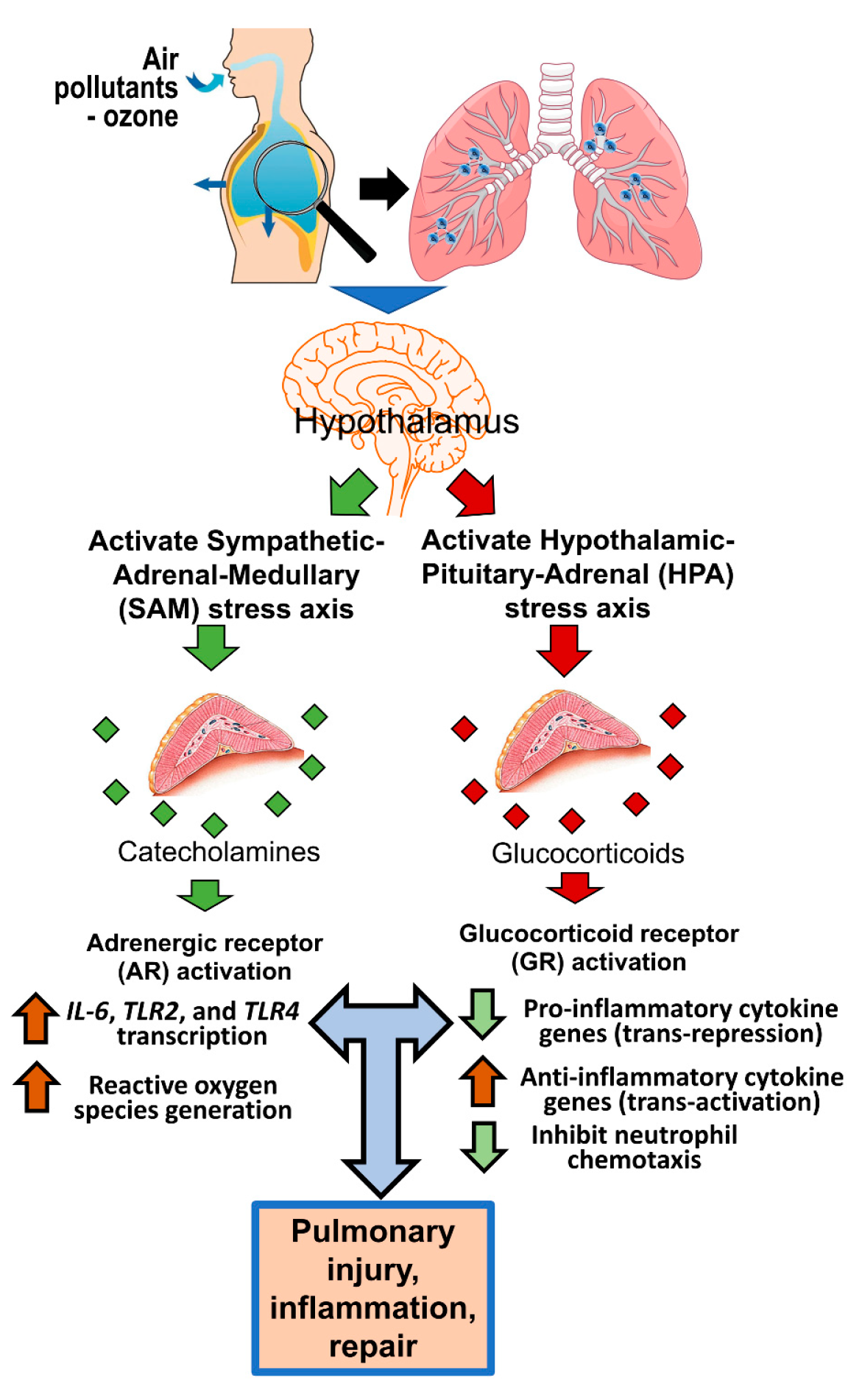
:1. Introduction
2. ARs and GRs, Their Subtypes, and Roles in Homeostatic Functions
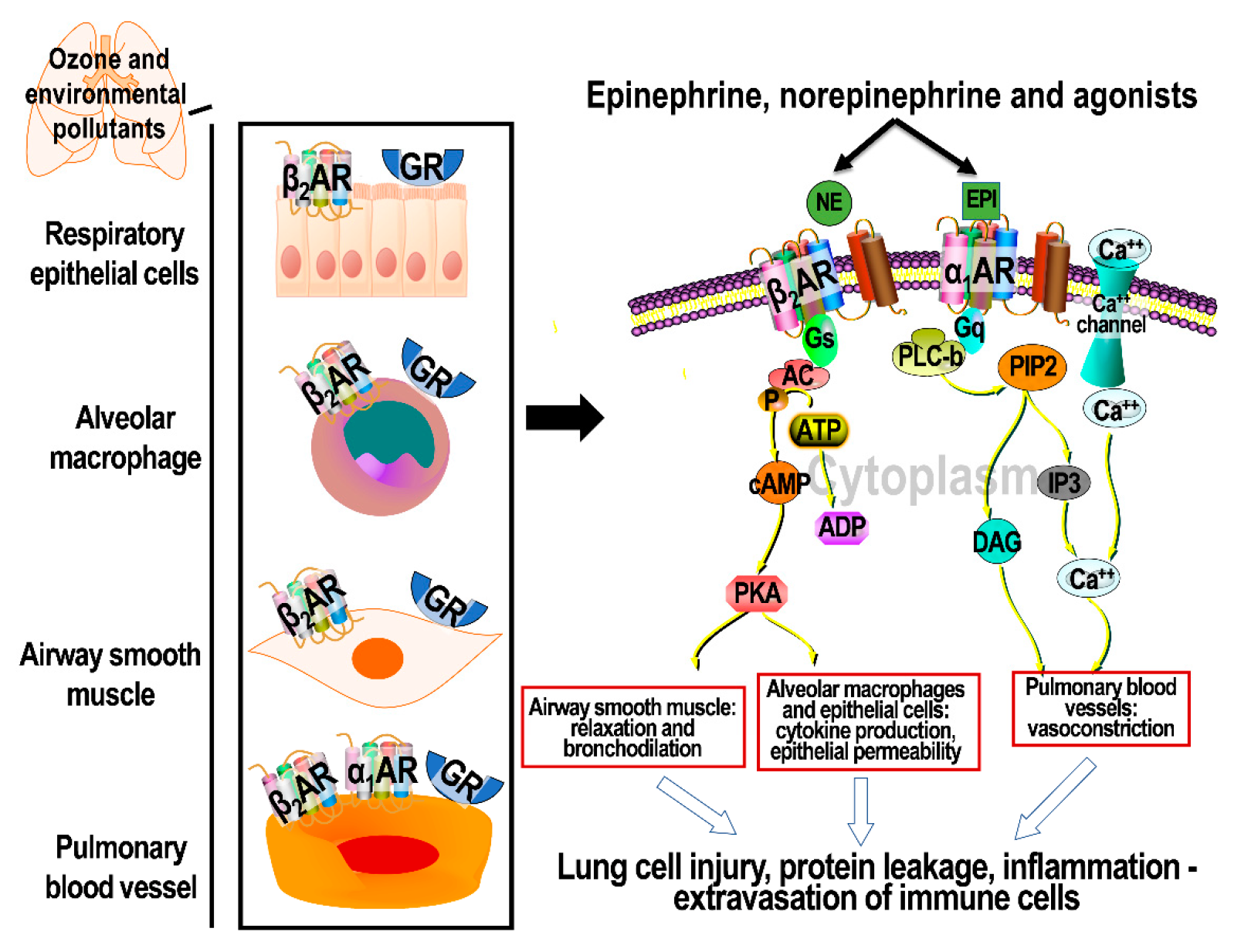
3. Cellular Signaling through Activation of ARs
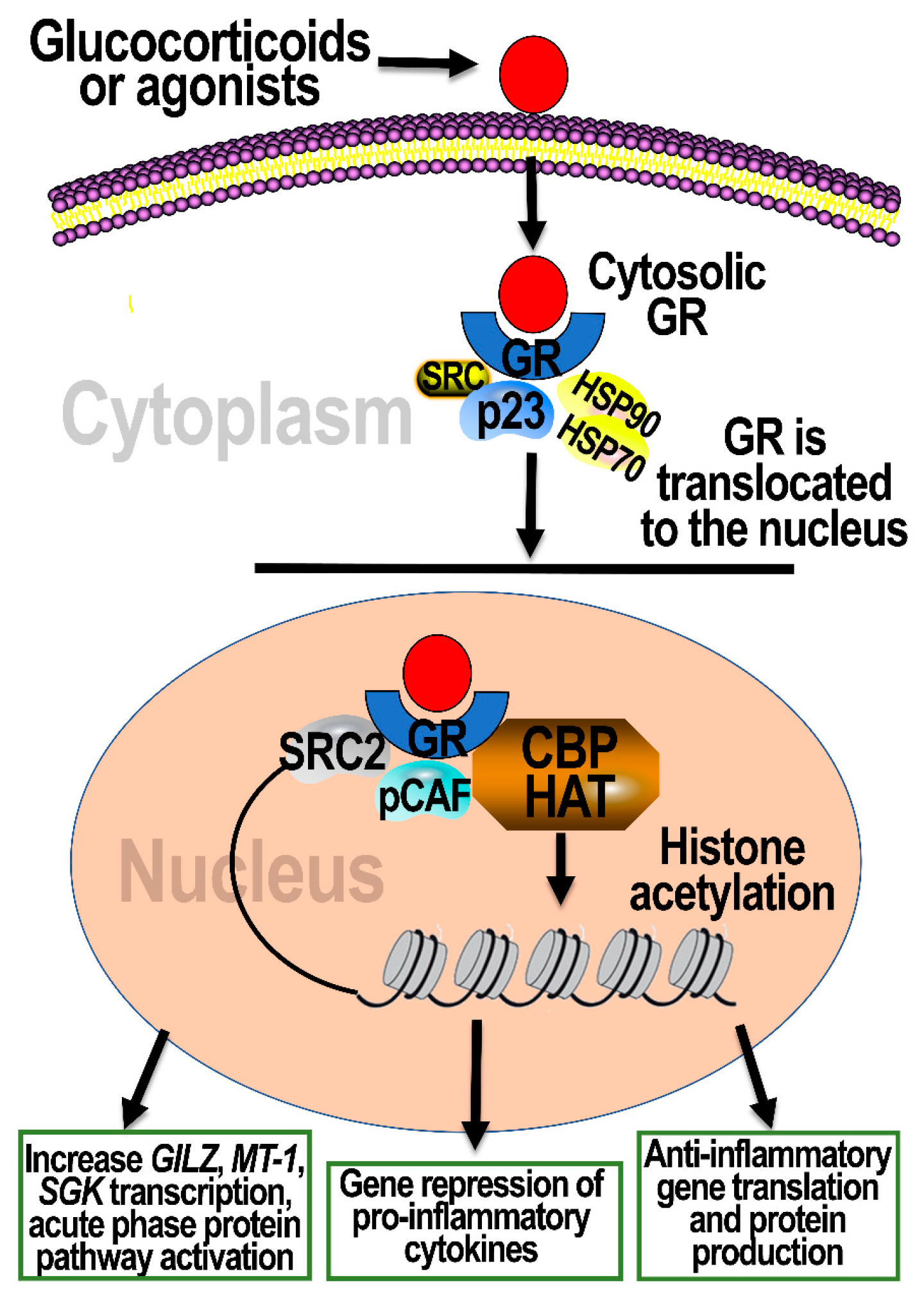
4. Cellular Signaling through Activation of GRs
5. Distribution of AR and GR Subtypes in the Lungs
6. ARs and GRs in Air-Pollutant-Induced Lung Injury and Inflammation
6.1. Air Pollution Studies Implicating the Role of ARs
6.2. Air Pollution Studies Implicating the Role of GRs
7. ARs and GRs in Ozone-Induced Lung Injury and Inflammation
7.1. Adrenalectomy Inhibits Lung Injury and Inflammation Induced by Acute Ozone Exposure
7.2. βAR and GR Activation Contribute to Ozone-Induced Lung Inflammation
7.3. AR and GR Antagonists Inhibit Ozone-Induced Lung Inflammation
7.4. The Role of βARs in Ozone-Induced Lung Protein Leakage
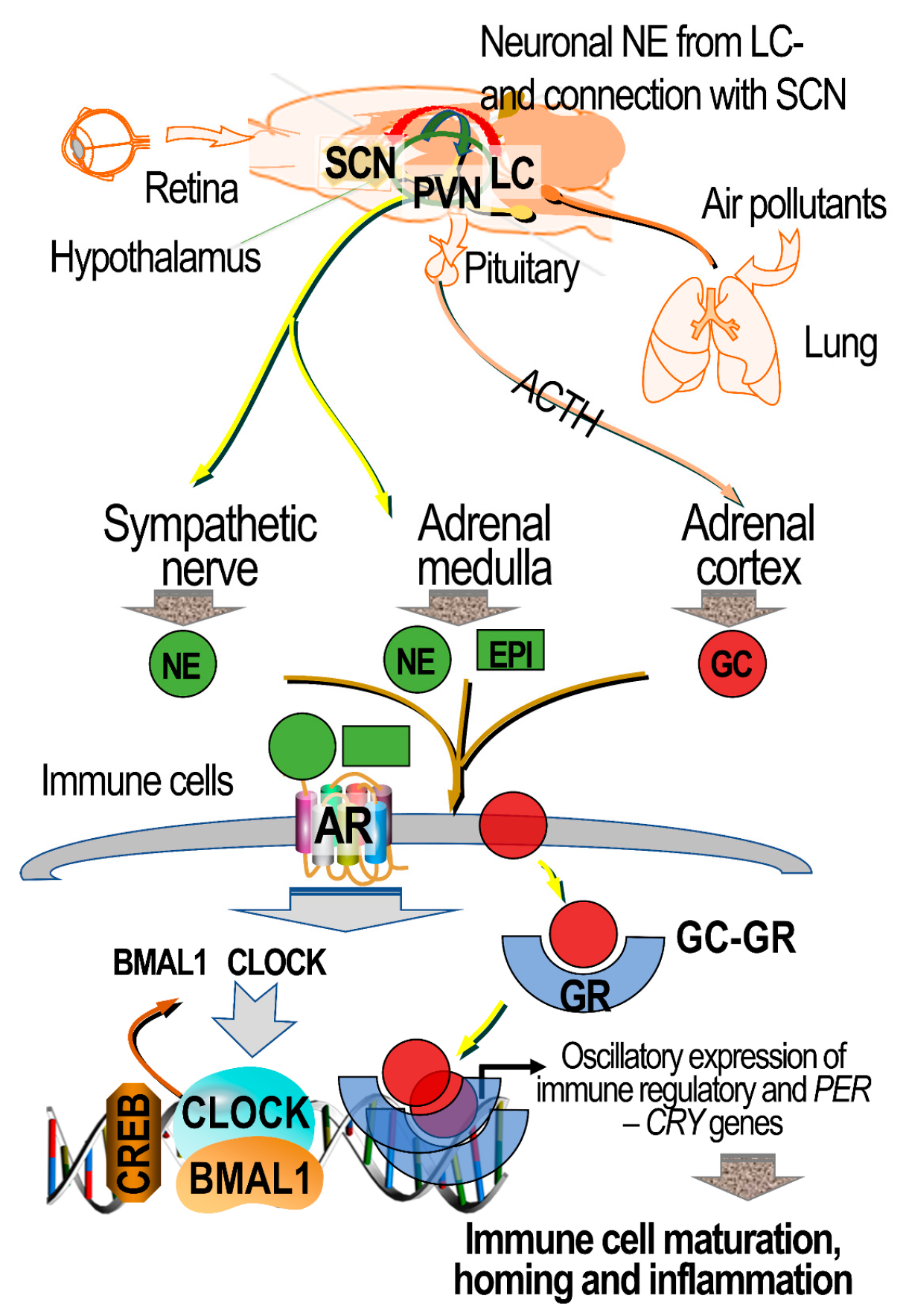
7.5. Circulating Ligands of ARs and GRs in Lung–Brain Communication
8. Potential Interactive Roles of ARs and GRs in Inflammatory Mechanisms
9. Air Pollution’s Impact on Circadian Clock Genes, and the Potential Mediating Roles of ARs and GRs
10. Research Gaps and Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Barnes, P.J. Distribution of Receptor Targets in the Lung. Proc. Am. Thorac. Soc. 2004, 1, 345–351. [Google Scholar] [CrossRef]
- Barnes, P.J. Glucocorticosteroids. Handb. Exp. Pharmacol. 2017, 237, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Brinchmann, B.C.; Le Ferrec, E.; Podechard, N.; Lagadic-Gossmann, M.; Holme, J.A.; Øvrevik, J. Organic chemicals from diesel exhaust particles affects intracellular calcium, inflammation and β-adrenoceptors in endothelial cells. Toxicol. Lett. 2019, 302, 18–27. [Google Scholar] [CrossRef]
- Henriquez, A.R.; Snow, S.J.; Schladweiler, M.C.; Miller, C.N.; Dye, J.; Ledbetter, A.D.; Richards, J.; Hargrove, M.M.; Williams, W.C.; Kodavanti, U.P. Beta-2 Adrenergic and Glucocorticoid Receptor Agonists Modulate Ozone-Induced Pulmonary Protein Leakage and Inflammation in Healthy and Adrenalectomized Rats. Toxicol. Sci. 2018, 166, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, A.R.; Snow, S.J.; Schladweiler, M.C.; Miller, C.N.; Dye, J.A.; Ledbetter, A.D.; Richards, J.E.; Mauge-Lewis, K.; McGee, M.A.; Kodavanti, U.P. Adrenergic and glucocorticoid receptor antagonists reduce ozone-induced lung injury and inflammation. Toxicol. Appl. Pharmacol. 2018, 339, 161–171. [Google Scholar] [CrossRef]
- Henriquez, A.R.; Snow, S.J.; Schladweiler, M.C.; Miller, C.N.; Dye, J.A.; Ledbetter, A.D.; Hargrove, M.M.; Richards, J.E.; Kodavanti, U.P. Exacerbation of ozone-induced pulmonary and systemic effects by β2-adrenergic and/or glucocorticoid receptor agonist/s. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, A.R.; House, J.S.; Snow, S.J.; Miller, C.N.; Schladweiler, M.C.; Fisher, A.; Ren, H.; Valdez, M.; Kodavanti, P.R.; Kodavanti, U.P. Ozone-Induced Dysregulation of Neuroendocrine Axes Requires Adrenal-Derived Stress Hormones. Toxicol. Sci. 2019, 172, 38–50. [Google Scholar] [CrossRef]
- Chiarella, S.E.; Soberanes, S.; Urich, D.; Morales-Nebreda, L.; Nigdelioglu, R.; Green, D.; Young, J.B.; Gonzalez, A.; Rosario, C.; Misharin, A.V.; et al. β2-Adrenergic agonists augment air pollution–induced IL-6 release and thrombosis. J. Clin. Investig. 2014, 124, 2935–2946. [Google Scholar] [CrossRef]
- Gao, Y.; Lv, J.; Lin, Y.; Li, X.; Wang, L.; Yin, Y.; Liu, Y. Effects ofβ-Adrenoceptor Subtypes on Cardiac Function in Myocardial Infarction Rats Exposed to Fine Particulate Matter (PM2.5). BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Zhou, T.; Zhong, Y.; Liao, J.; Wang, G.; Li, X.; Qian, X.; Xiang, P.; Chen, X.; Xu, Z.; Zhang, F.; et al. A prospective study of salvational intervention with ICS/LABA for reducing chronic obstructive pulmonary disease exacerbation under severe air pollution (SIRCAP) in Beijing: Protocol of a multi-center randomized controlled trial. BMC Pulm. Med. 2019, 19, 22. [Google Scholar] [CrossRef]
- Rabe, K.F.; Martinez, F.J.; Ferguson, G.T.; Wang, C.; Singh, D.; Wedzicha, J.A.; Trivedi, R.; Rose, E.S.; Ballal, S.; McLaren, J.; et al. Triple Inhaled Therapy at Two Glucocorticoid Doses in Moderate-to-Very-Severe COPD. N. Engl. J. Med. 2020, 383, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Beasley, R.; Holliday, M.; Reddel, H.; Braithwaite, I.; Ebmeier, S.; Hancox, R.J.; Harrison, T.; Houghton, C.; Oldfield, K.; Papi, A.; et al. Controlled Trial of Budesonide–Formoterol as Needed for Mild Asthma. N. Engl. J. Med. 2019, 380, 2020–2030. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.B.; Karoly, E.D.; Jones, J.C.; Ward, W.O.; Vallanat, B.D.; Andrews, D.L.; Schladweiler, M.C.; Snow, S.J.; Bass, V.L.; Richards, J.E.; et al. Inhaled ozone (O3)-induces changes in serum metabolomic and liver transcriptomic profiles in rats. Toxicol. Appl. Pharmacol. 2015, 286, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, A.R.; Snow, S.J.; Schladweiler, M.C.; Miller, C.N.; Kodavanti, U.P. Independent roles of beta-adrenergic and glucocorticoid receptors in systemic and pulmonary effects of ozone. Inhal. Toxicol. 2020, 32, 155–169. [Google Scholar] [CrossRef]
- De Kloet, E.R.; De Kloet, S.F.; De Kloet, C.S.; De Kloet, A.D. Top-down and bottom-up control of stress-coping. J. Neuroendocr. 2018, 31, e12675. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; Nawreen, N.; Smail, M.A.; Cotella, E.M. Brain mechanisms of HPA axis regulation: Neurocircuitry and feedback in context Richard Kvetnansky lecture. Stress 2020, 23, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Mracsko, E.; Liesz, A.; Karcher, S.; Zorn, M.; Bari, F.; Veltkamp, R. Differential effects of sympathetic nervous system and hypothalamic–pituitary–adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav. Immun. 2014, 41, 200–209. [Google Scholar] [CrossRef]
- Herman, J.P. Regulation of Hypothalamo-Pituitary-Adrenocortical Responses to Stressors by the Nucleus of the Solitary Tract/Dorsal Vagal Complex. Cell. Mol. Neurobiol. 2018, 38, 25–35. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Giridharan, T.; Macdonald, C.R.; Hylander, B.L.; Repasky, E.A. An overview of the role of sympathetic regulation of immune responses in infectious disease and autoimmunity. Int. J. Hyperth. 2018, 34, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mravec, B.; Vargovic, P.; Filipcik, P.; Novak, M.; Kvetnansky, R. Effect of a single and repeated stress exposure on gene expression of catecholamine biosynthetic enzymes in brainstem catecholaminergic cell groups in rats. Eur. J. Neurosci. 2015, 42, 1872–1886. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.B.; Thayer, J.F.; Vedhara, K. Stress and Health: A Review of Psychobiological Processes. Annu. Rev. Psychol. 2021, 72, 663–688. [Google Scholar] [CrossRef]
- Kolodkin, A.; Sahin, N.; Phillips, A.; Hood, S.R.; Bruggeman, F.J.; Westerhoff, H.; Plant, N. Optimization of stress response through the nuclear receptor-mediated cortisol signalling network. Nat. Commun. 2013, 4, 1792. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Cui, S.; Cui, X.; Liu, Y.; Hu, X.; Zhao, H.; Qin, Y.; Kurban, N.; Zhang, Y. Anti-stress effects of combined glucocorticoid and mineralocorticoid receptor blockade in the paraventricular nucleus of the hypothalamus. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Joëls, M.; Sarabdjitsingh, R.A.; Karst, H. Unraveling the Time Domains of Corticosteroid Hormone Influences on Brain Activity: Rapid, Slow, and Chronic Modes. Pharmacol. Rev. 2012, 64, 901–938. [Google Scholar] [CrossRef] [PubMed]
- Dietl, H.; Prast, H.; Philippu, A. Pulsatile release of catecholamines in the hypothalamus of conscious rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1993, 347, 28–33. [Google Scholar] [CrossRef]
- Molinoff, P.B. Alpha- and beta-Adrenergic Receptor Subtypes Properties, Distribution and Regulation. Drugs 1984, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.N.; Cassagnol, M. Alpha Adrenergic Receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539830/ (accessed on 12 April 2021).
- Alhayek, S.; Preuss, C.V. Beta 1 Receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532904/ (accessed on 4 December 2020).
- Yang, L.-K.; Tao, Y.-X. Physiology and pathophysiology of the β3-adrenergic receptor. Prog. Mol. Biol. Transl. Sci. 2019, 161, 91–112. [Google Scholar] [CrossRef]
- Rang, H.P.; Ritter, J.M.; Flower, R.J.; Henderson, G. Rang and Dale’s Pharmacology, 8th ed.; Elsevier: Oxford, UK, 2016; p. 179. ISBN 9780702053627. [Google Scholar]
- Tanaka, Y.; Horinouchi, T.; Koike, K. New insights into β-adrenoceptors in smooth muscle: Distribution of receptor subtypes and molecular mechanisms triggering muscle relaxation. Clin. Exp. Pharmacol. Physiol. 2005, 32, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Liu, W.; Schlenk, D.; Liu, J. Glucocorticoid and mineralocorticoid receptors and corticosteroid homeostasis are potential targets for endocrine-disrupting chemicals. Environ. Int. 2019, 133, 105133. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.H.; Cormack, J.; Mallinson, D.; Mutungi, G. A membrane glucocorticoid receptor mediates the rapid/non-genomic actions of glucocorticoids in mammalian skeletal muscle fibres. J. Physiol. 2013, 591, 5171–5185. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Weinshenker, D. Adrenaline Rush: The Role of Adrenergic Receptors in Stimulant-Induced Behaviors. Mol. Pharmacol. 2014, 85, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecasteele, G.; Eschenhagen, T.; Scholz, H.; Stein, B.; Verde, I.; Fischmeister, R. Muscarinic and β-adrenergic regulation of heart rate, force of contraction and calcium current is preserved in mice lacking endothelial nitric oxide synthase. Nat. Med. 1999, 5, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.; Van Breemen, C. Role of intracellular Ca2+ sequestration in β-adrenergic relaxation of a smooth muscle. Nat. Cell Biol. 1979, 281, 682–683. [Google Scholar] [CrossRef]
- Latorre, G.; Cardenas, V.; Lopez, O. Mechanism of the effect of constant infusion of epinephrine on blood pressure, heart rate and arterial hematocrit in normal and sympathectomized-splanchnicectomized dogs. Arch. Int. Physiol. Biochim. 1960, 68, 785–792. [Google Scholar] [CrossRef]
- El-Merahbi, R.; Viera, J.T.; Valdes, A.L.; Kolczynska, K.; Reuter, S.; Löffler, M.C.; Erk, M.; Ade, C.P.; Karwen, T.; Mayer, A.E.; et al. The adrenergic-induced ERK3 pathway drives lipolysis and suppresses energy dissipation. Genes Dev. 2020, 34, 495–510. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.J. The ins and outs of adrenergic signaling. J. Mol. Med. 2015, 93, 955–962. [Google Scholar] [CrossRef]
- Vass, M.; Kooistra, A.; Yang, D.; Stevens, R.C.; Wang, M.-W.; de Graaf, C. Chemical Diversity in the G Protein-Coupled Receptor Superfamily. Trends Pharmacol. Sci. 2018, 39, 494–512. [Google Scholar] [CrossRef]
- Bahouth, S.W.; Nooh, M.M. Barcoding of GPCR trafficking and signaling through the various trafficking roadmaps by compartmentalized signaling networks. Cell. Signal. 2017, 36, 42–55. [Google Scholar] [CrossRef]
- Singh, M.; Moniri, N.H. Reactive oxygen species are required for β2 adrenergic receptor–β-arrestin interactions and signaling to ERK1/2. Biochem. Pharmacol. 2012, 84, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of Receptor Fate by Ubiquitination of Activated beta 2-Adrenergic Receptor and beta-Arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef]
- Lin, Y.; Smrcka, A.V. Understanding Molecular Recognition by G protein βγ Subunits on the Path to Pharmacological Targeting. Mol. Pharmacol. 2011, 80, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, D.L.; Hutchinson, D.S.; Bengtsson, T. Beta(2)-Adrenergic activation increases glycogen synthesis in L6 skeletal muscle cells through a signalling pathway independent of cyclic AMP. Diabetologia 2007, 50, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Meitzen, J.; Luoma, J.I.; Stern, C.M.; Mermelstein, P.G. β1-Adrenergic receptors activate two distinct signaling pathways in striatal neurons. J. Neurochem. 2010, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.J.; Hofmann, K.P. Spatial and Temporal Aspects of Signaling by G-Protein–Coupled Receptors. Mol. Pharmacol. 2015, 88, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Foguet, M.; Lübbert, H.; Stühmer, W. Ca2+ oscillations and Ca2+ influx in Xenopus oocytes expressing a novel 5-hydroxytryptamine receptor. J. Physiol. 1993, 469, 653–671. [Google Scholar] [CrossRef] [PubMed]
- Thomson, L.M.; Raven, P.W.; Smith, K.E.; Hinson, J.P. Effects of metyrapone on hepatic cortisone-cortisol conversion in the rat. Endocr. Res. 1998, 24, 607–611. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Cidlowski, J.A. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef]
- Yeager, M.P.; Pioli, P.A.; Guyre, P.M. Cortisol Exerts bi-phasic Regulation of Inflammation in Humans. Dose-Response 2010, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Van Looveren, K.; Van Boxelaere, M.; Callaerts-Vegh, Z.; Libert, C. Cognitive dysfunction in mice lacking proper glucocorticoid receptor dimerization. PLoS ONE 2019, 14, e0226753. [Google Scholar] [CrossRef] [Green Version]
- Widén, C.; Gustafsson, J.-A.; Wikström, A.-C. Cytosolic glucocorticoid receptor interaction with nuclear factor-kappaB proteins in rat liver cells. Biochem. J. 2003, 373, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unlap, T.; Jope, R.S. Inhibition of NFkB DNA binding activity by glucocorticoids in rat brain. Neurosci. Lett. 1995, 198, 41–44. [Google Scholar] [CrossRef]
- Ayroldi, E.; Cannarile, L.; Migliorati, G.; Nocentini, G.; Delfino, D.V.; Riccardi, C. Mechanisms of the anti-inflammatory effects of glucocorticoids: Genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012, 26, 4805–4820. [Google Scholar] [CrossRef]
- Yang, Y.H.; Aeberli, D.; Dacumos, A.; Xue, J.R.; Morand, E. Annexin-1 Regulates Macrophage IL-6 and TNF via Glucocorticoid-Induced Leucine Zipper. J. Immunol. 2009, 183, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.-Y.; Wu, S.-C.; Yao, C.-L.; Hsu, C.-N.; Jen, C.-Y.; Chen, Y.-H. Glucocorticoid transiently upregulates mitochondrial biogenesis in the osteoblast. Chin. J. Physiol. 2020, 63, 286. [Google Scholar] [CrossRef]
- Hampl, R.; Vondra, K. Peripheral Sensitivity to Steroids Revisited. Physiol. Res. 2017, 66, S295–S303. [Google Scholar] [CrossRef]
- Muzikar, K.A.; Nickols, N.G.; Dervan, P.B. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 16598–16603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, P.W.; Magness, R.R.; Muntz, K.H.; DeBeltz, D.; Buja, L.M. Alpha 1-adrenergic receptors in pulmonary and systemic vascular smooth muscle. Alterations with development and pregnancy. Circ. Res. 1990, 67, 1193–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Beta-adrenergic receptors and their regulation. Am. J. Respir. Crit. Care Med. 1995, 152, 838–860. [Google Scholar] [CrossRef] [PubMed]
- Henry, P.J.; Rigby, P.J.; Goldie, R.G. Distribution of beta 1- and beta 2-adrenoceptors in mouse trachea and lung: A quantitative autoradiographic study. Br. J. Pharmacol. 1990, 99, 136–144. [Google Scholar] [CrossRef]
- Downs, C.A.; Kriener, L.H.; Yu, L.; Eaton, U.C.; Jain, L.; Helms, M.N. β-Adrenergic agonists differentially regulate highly selective and nonselective epithelial sodium channels to promote alveolar fluid clearance in vivo. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L1167–L1178. [Google Scholar] [CrossRef] [PubMed]
- Factor, P.; Adir, Y.; Mutlu, G.M.; Burhop, J.; Dumasius, V. Effects of β2-adrenergic receptor overexpression on alveolar epithelial active transport. J. Allergy Clin. Immunol. 2002, 110, S242–S246. [Google Scholar] [CrossRef] [PubMed]
- Bossard, F.; Silantieff, E.; Lavazais-Blancou, E.; Robay, A.; Sagan, C.; Rozec, B.; Gauthier, C. β1, β2, and β3 Adrenoceptors and Na+/H+Exchanger Regulatory Factor 1 Expression in Human Bronchi and Their Modifications in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 44, 91–98. [Google Scholar] [CrossRef]
- Gu, Q.; Lin, Y.S.; Lee, L.Y. Epinephrine enhances the sensitivity of rat vagal chemosensitive neurons: Role of beta3-adrenoceptor. J. Appl. Physiol. 2007, 102, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- García-Álvarez, A.; Pereda, D.; García-Lunar, I.; Sanz-Rosa, D.; Fernández-Jiménez, R.; García-Prieto, J.; Nuño-Ayala, M.; Sierra, F.; Santiago, E.; Sandoval, E.; et al. Beta-3 adrenergic agonists reduce pulmonary vascular resistance and improve right ventricular performance in a porcine model of chronic pulmonary hypertension. Basic Res. Cardiol. 2016, 111, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, G.; Chen, Z.; Fang, X.; Liu, M.; Hao, G.; An, H.; Zhang, Z.; Lu, L.; Zhang, J.; Zhang, L. Relationship between the Autoantibody and Expression of β3-Adrenoceptor in Lung and Heart. PLoS ONE 2013, 8, e68747. [Google Scholar] [CrossRef] [Green Version]
- Schwiebert, L.M.; Stellato, C.; Schleimer, R.P. The Epithelium as a Target of Glucocorticoid Action in the Treatment of Asthma. Am. J. Respir. Crit. Care Med. 1996, 154, S16–S20. [Google Scholar] [CrossRef] [PubMed]
- Hamid, Q. Effects of steroids on inflammation and cytokine gene expression in airway inflammation. J. Allergy Clin. Immunol. 2003, 112, 636–638. [Google Scholar] [CrossRef]
- Stolfo, D.; Uijl, A.; Benson, L.; Schrage, B.; Fudim, M.; Asselbergs, F.W.; Koudstaal, S.; Sinagra, G.; Dahlström, U.; Rosano, G.; et al. Association between beta-blocker use and mortality/morbidity in older patients with heart failure with reduced ejection fraction. A propensity score-matched analysis from the Swedish Heart Failure Registry. Eur. J. Heart Fail. 2020, 22, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Sun, Y.; Ding, N.; Lu, L.; Chen, Y. Beta-Blockers Reduced the Risk of Mortality and Exacerbation in Patients with COPD: A Meta-Analysis of Observational Studies. PLoS ONE 2014, 9, e113048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.K.; Sorrentino, M.J. Beta blockers for CHF. Adrenergic blockade dramatically reduces morbidity and mortality. Postgrad. Med. 2001, 109, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Glucocorticoid signaling in the heart: A cardiomyocyte perspective. J. Steroid Biochem. Mol. Biol. 2015, 153, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Therapeutic approaches to asthma–chronic obstructive pulmonary disease overlap syndromes. J. Allergy Clin. Immunol. 2015, 136, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Li, F.; Ryffel, B.; Togbe, D.; Chung, K.F. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Bromberg, P.A. Mechanisms of the acute effects of inhaled ozone in humans. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 2771–2781. [Google Scholar] [CrossRef]
- Kodavanti, U.P. Stretching the stress boundary: Linking air pollution health effects to a neurohormonal stress response. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 2880–2890. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, U.P. Susceptibility Variations in Air Pollution Health Effects: Incorporating Neuroendocrine Activation. Toxicol. Pathol. 2019, 47, 962–975. [Google Scholar] [CrossRef]
- Allen, J.L.; Liu, X.; Pelkowski, S.; Palmer, B.; Conrad, K.; Oberdörster, G.; Weston, D.; Mayer-Pröschel, M.; Cory-Slechta, D.A. Early Postnatal Exposure to Ultrafine Particulate Matter Air Pollution: Persistent Ventriculomegaly, Neurochemical Disruption, and Glial Activation Preferentially in Male Mice. Environ. Health Perspect. 2014, 122, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Greve, H.J.; Mumaw, C.L.; Messenger, E.J.; Kodavanti, P.R.S.; Royland, J.L.; Kodavanti, U.P.; Block, M.L. Diesel exhaust impairs TREM2 to dysregulate neuroinflammation. J. Neuroinflamm. 2020, 17, 351. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, A.R.; Williams, W.; Snow, S.J.; Schladweiler, M.C.; Fisher, C.; Hargrove, M.M.; Alewel, D.I.; Colonna, C.H.; Gavette, S.H.; Miller, C.N.; et al. The dynamicity of acute ozone-induced systemic immune response. Toxicology 2021, 458, 152823. [Google Scholar] [CrossRef] [PubMed]
- Torjussen, T.M.; Munthe-Kaas, M.C.; Mowinckel, P.; Carlsen, K.-H.; Undlien, D.; Carlsen, K.C.L. Childhood lung function and the association with β2-adrenergic receptor haplotypes. Acta Paediatr. 2013, 102, 727–731. [Google Scholar] [CrossRef]
- Zhang, G.; Hayden, C.M.; Khoo, S.-K.; Candelaria, P.; Laing, I.A.; Turner, S.; Franklin, P.; Stick, S.; Landau, L.; Goldblatt, J.; et al. 2-Adrenoceptor polymorphisms and asthma phenotypes: Interactions with passive smoking. Eur. Respir. J. 2007, 30, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, C.R.; Wellenius, G.A.; Diaz, E.A.; Lawrence, J.; Coull, B.A.; Akiyama, I.; Lee, L.M.; Okabe, K.; Verrier, R.L.; Godleski, J.J. Mechanisms of Inhaled Fine Particulate Air Pollution–Induced Arterial Blood Pressure Changes. Environ. Health Perspect. 2009, 117, 361–366. [Google Scholar] [CrossRef]
- Li, H.; Cai, J.; Chen, R.; Zhao, Z.; Ying, Z.; Wang, L.; Chen, J.; Hao, K.; Kinney, P.L.; Chen, H.; et al. Particulate Matter Exposure and Stress Hormone Levels: A Randomized, Double-Blind, Crossover Trial of Air Purification. Circulation 2017, 136, 618–627. [Google Scholar] [CrossRef]
- Hajat, A.; Hazlehurst, M.F.; Golden, S.H.; Merkin, S.S.; Seeman, T.; Szpiro, A.A.; Kaufman, J.D.; Roux, A.D. The cross-sectional and longitudinal association between air pollution and salivary cortisol: Evidence from the Multi-Ethnic Study of Atherosclerosis. Environ. Int. 2019, 131, 105062. [Google Scholar] [CrossRef]
- Hajat, A.; Roux, A.V.D.; Castro-Diehl, C.; Cosselman, K.; Golden, S.H.; Hazlehurst, M.F.; Szpiro, A.; Vedal, S.; Kaufman, J. The Association between Long-Term Air Pollution and Urinary Catecholamines: Evidence from the Multi-Ethnic Study of Atherosclerosis. Environ. Health Perspect. 2019, 127, 057007. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Wu, B.; Tan, C.; He, X.; Xu, B.; Yu, G.; Wang, H. β-Arrestin2 Inhibits Expression of Inflammatory Cytokines in BEAS-2B Lung Epithelial Cells Treated with Cigarette Smoke Condensate via Inhibition of Autophagy. Cell. Physiol. Biochem. 2018, 50, 1270–1285. [Google Scholar] [CrossRef]
- Kizaki, T.; Shirato, K.; Sakurai, T.; Ogasawara, J.E.; Oh-ishi, S.; Matsuoka, T.; Izawa, T.; Imaizumi, K.; Haga, S.; Ohno, H. Beta2-adrenergic receptor regulate Toll-like receptor 4-induced late-phase NF-kappaB activation. Mol. Immunol. 2009, 46, 1195–1203. [Google Scholar] [CrossRef]
- Snow, S.J.; McGee, M.A.; Henriquez, A.; Richards, J.E.; Schladweiler, M.C.; Ledbetter, A.D.; Kodavanti, U.P. Respiratory Effects and Systemic Stress Response Following Acute Acrolein Inhalation in Rats. Toxicol. Sci. 2017, 158, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Asico, L.D.; Zanos, P.; Mahabeleshwar, G.H.; Gangwar, R.S.; Xia, C.; Duan, L.; Cisse, Y.-M.; Rengasamy, P.; A Jose, P.; et al. Alpha2B-Adrenergic Receptor Overexpression in the Brain Potentiate Air Pollution-induced Behavior and Blood Pressure Changes. Toxicol. Sci. 2019, 169, 95–107. [Google Scholar] [CrossRef]
- Knuckles, T.L.; Stapleton, P.A.; Minarchick, V.C.; Esch, L.; McCAWLEY, M.; Hendryx, M.; Nurkiewicz, T.R. Air Pollution Particulate Matter Collected from an Appalachian Mountaintop Mining Site Induces Microvascular Dysfunction. Microcirculation 2012, 20, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Z.; Wei, Y.; Li, X.; Yang, L.; Liu, H.; Guo, C.; Zhang, L.; Li, N.; Guo, S.; Qian, Y.; et al. Exposure to Ambient Air Particles Increases the Risk of Mental Disorder: Findings from a Natural Experiment in Beijing. Int. J. Environ. Res. Public Health 2018, 15, 160. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, Y.; Liu, W.; Liu, J. Potential endocrine-disrupting effects of metals via interference with glucocorticoid and mineralocorticoid receptors. Environ. Pollut. 2018, 242, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.M.; Vladisavljevic, D.; Mohottalage, S.; Kumarathasan, P.; Vincent, R. Mapping Acute Systemic Effects of Inhaled Particulate Matter and Ozone: Multiorgan Gene Expression and Glucocorticoid Activity. Toxicol. Sci. 2013, 135, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Dimitrov, S.; Cheng, T.; Redwine, L.; Pruitt, C.; Mills, P.J.; Ziegler, M.G.; Green, J.M.; Shaikh, F.; Wilson, K. Beta-adrenergic receptor mediated inflammation control by monocytes is associated with blood pressure and risk factors for cardiovascular disease. Brain Behav. Immun. 2015, 50, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kox, M.; van Eijk, L.T.; Zwaag, J.; Wildenberg, J.V.D.; Sweep, F.; van der Hoeven, J.G.; Pickkers, P. Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7379–7384. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, S.; Brestoff, J.R.; Flamar, A.-L.; Moeller, J.B.; Klose, C.S.N.; Rankin, L.C.; Yudanin, N.A.; Monticelli, L.A.; Putzel, G.G.; Rodewald, H.-R.; et al. β2-adrenergic receptor–mediated negative regulation of group 2 innate lymphoid cell responses. Science 2018, 359, 1056–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.P.; Al-Sawalha, N.A.; Parra, S.; Pokkunuri, I.; Omoluabi, O.; Okulate, A.A.; Li, E.W.; Hazen, M.; Gonzalez-Granado, J.M.; Daly, C.J.; et al. β2-Adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility. Proc. Natl. Acad. Sci. USA 2017, 114, E9163–E9171. [Google Scholar] [CrossRef] [Green Version]
- Lorton, D.; Bellinger, D.L. Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef] [Green Version]
- Bass, V.; Gordon, C.; Jarema, K.; MacPhail, R.; Cascio, W.; Phillips, P.; Ledbetter, A.; Schladweiler, M.; Andrews, D.; Miller, D.; et al. Ozone induces glucose intolerance and systemic metabolic effects in young and aged brown Norway rats. Toxicol. Appl. Pharmacol. 2013, 273, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, A.I.; Singanayagam, A.; Wiater, E.; Edwards, M.R.; Montminy, M.; Johnston, S.L. β2-Agonists Enhance Asthma-Relevant Inflammatory Mediators in Human Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 128–132. [Google Scholar] [CrossRef]
- Kurhanewicz, N.; Ledbetter, A.; Farraj, A.; Hazari, M. TRPA1 mediates the cardiac effects of acrolein through parasympathetic dominance but also sympathetic modulation in mice. Toxicol. Appl. Pharmacol. 2018, 347, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Rankin, G.D.; Kabéle, M.; Brown, R.; Macefield, V.G.; Sandström, T.; Bosson, J.A. Acute Exposure to Diesel Exhaust Increases Muscle Sympathetic Nerve Activity in Humans. J. Am. Hear. Assoc. 2021, 18, e018448. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and resistance to inhaled glucocorticoids: Evidence, mechanisms and gaps to fill. Pharmacol. Ther. 2019, 194, 1–21. [Google Scholar] [CrossRef]
- Miller, D.B.; Ghio, A.J.; Karoly, E.D.; Bell, L.N.; Snow, S.J.; Madden, M.C.; Soukup, J.M.; Cascio, W.E.; Gilmour, M.I.; Kodavanti, U.P. Ozone Exposure Increases Circulating Stress Hormones and Lipid Metabolites in Humans. Am. J. Respir. Crit. Care Med. 2016, 193, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Snow, S.J.; Henriquez, A.R.; Costa, D.L.; Kodavanti, U.P. Neuroendocrine Regulation of Air Pollution Health Effects: Emerging Insights. Toxicol. Sci. 2018, 164, 9–20. [Google Scholar] [CrossRef]
- Kodavanti, U.P.; Ledbetter, A.D.; Thomas, R.F.; Richards, J.E.; Ward, W.O.; Schladweiler, M.C.; Costa, D.L. Variability in ozone-induced pulmonary injury and inflammation in healthy and cardiovascular-compromised rat models. Inhal. Toxicol. 2015, 27, 39–53. [Google Scholar] [CrossRef]
- Miller, D.B.; Snow, S.J.; Schladweiler, M.C.; Richards, J.E.; Ghio, A.J.; Ledbetter, A.D.; Kodavanti, U.P. Acute Ozone-Induced Pulmonary and Systemic Metabolic Effects Are Diminished in Adrenalectomized Rats. Toxicol. Sci. 2016, 150, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriquez, A.; House, J.; Miller, D.B.; Snow, S.J.; Fisher, A.; Ren, H.; Schladweiler, M.C.; Ledbetter, A.D.; Wright, F.; Kodavanti, U.P. Adrenal-derived stress hormones modulate ozone-induced lung injury and inflammation. Toxicol. Appl. Pharmacol. 2017, 329, 249–258. [Google Scholar] [CrossRef]
- Thomas, J.; Stalker, A.; Breznan, D.; Thomson, E.M. Ozone-dependent increases in lung glucocorticoids and macrophage response: Effect modification by innate stress axis function. Environ. Toxicol. Pharmacol. 2021, 86, 103662. [Google Scholar] [CrossRef]
- Gent, J.F.; Triche, E.W.; Holford, T.R.; Belanger, K.; Bracken, M.B.; Beckett, W.S.; Leaderer, B.P. Association of Low-Level Ozone and Fine Particles With Respiratory Symptoms in Children With Asthma. JAMA 2003, 290, 1859. [Google Scholar] [CrossRef] [Green Version]
- Thomson, E.M.; Filiatreault, A.; Guénette, J. Stress hormones potential mediators of air pollutant effects on the brain: Rapid induction of glucocorticoid-responsive genes. Environ. Res. 2019, 178, 108717. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Haas, J.; Panettieri, R.A., Jr.; Johnson, M.; Johnston, S.L. Corticosteroids and beta2 agonists differentially regulate rhinovirus-induced interleukin-6 via distinct Cis-acting elements. J. Biol. Chem. 2007, 282, 15366–15375. [Google Scholar] [CrossRef] [Green Version]
- Dorotea, D.; Ha, H. Activation of β2 adrenergic receptor signaling modulates inflammation: A target limiting the progression of kidney diseases. Arch. Pharmacal Res. 2021, 44, 49–62. [Google Scholar] [CrossRef]
- Baluk, P.; McDonald, D.M. The beta 2-adrenergic receptor agonist formoterol reduces microvascular leakage by inhibiting endothelial gap formation. Am. J. Physiol. Cell. Mol. Physiol. 1994, 266, L461–L468. [Google Scholar] [CrossRef]
- Wagner, J.G.; Allen, K.; Yang, H.-Y.; Nan, B.; Morishita, M.; Mukherjee, B.; Dvonch, J.T.; Spino, C.; Fink, G.D.; Rajagopalan, S.; et al. Cardiovascular Depression in Rats Exposed to Inhaled Particulate Matter and Ozone: Effects of Diet-Induced Metabolic Syndrome. Environ. Heal. Perspect. 2014, 122, 27–33. [Google Scholar] [CrossRef]
- Bölter, C.; Gabriel, P.; Appelt, P.; Salameh, A.; Schierle, K.; Rassler, B. Effects of Adrenergic Agonists and Antagonists on Cardiopulmonary Function During Normobaric Hypoxia in Rat. Front. Physiol. 2019, 10, 860. [Google Scholar] [CrossRef]
- Raßler, B.; Reißig, C.; Briest, W.; Tannapfel, A.; Zimmer, H.-G. Catecholamine-induced pulmonary edema and pleural effusion in rats—α- and β-adrenergic effects. Respir. Physiol. Neurobiol. 2003, 135, 25–37. [Google Scholar] [CrossRef]
- Rassler, B. Role of α- and β-adrenergic Mechanisms in the Pathogenesis of Pulmonary Injuries Characterized by Edema, Inflammation and Fibrosis. Cardiovasc. Hematol. Disord. Targets 2014, 13, 197–207. [Google Scholar] [CrossRef]
- Gackière, F.; Saliba, L.; Baude, A.; Bosler, O.; Strube, C. Ozone inhalation activates stress-responsive regions of the CNS. J. Neurochem. 2011, 117, 961–972. [Google Scholar] [CrossRef]
- Mostafa, M.M.; Rider, C.F.; Wathugala, N.D.; Leigh, R.; Giembycz, M.A.; Newton, R. Transcriptome-level interactions between budesonide and formoterol provide insight into the mechanism of action of ICS/LABA combination therapy in asthma. Mol. Pharmacol. 2020, 99, 197–216. [Google Scholar] [CrossRef]
- Rider, C.F.; Altonsy, M.O.; Mostafa, M.; Shah, S.V.; Sasse, S.; Manson, M.L.; Yan, D.; Kärrman-Mårdh, C.; Miller-Larsson, A.; Gerber, A.N.; et al. Long-Acting β2-Adrenoceptor Agonists Enhance Glucocorticoid Receptor (GR)–Mediated Transcription by Gene-Specific Mechanisms Rather Than Generic Effects via GR. Mol. Pharmacol. 2018, 94, 1031–1046. [Google Scholar] [CrossRef] [Green Version]
- Pilipović, I.; Radojević, K.; Perišić, M.; Leposavić, G. Glucocorticoid-catecholamine interplay within the composite thymopoietic regulatory network. Ann. N. Y. Acad. Sci. 2012, 1261, 34–41. [Google Scholar] [CrossRef]
- Lucki, A.; Klein, E.; Karry, R.; Ben-Shachar, D. Dexamethasone in the presence of desipramine enhances MAPK/ERK1/2 signaling possibly via its interference with β-arrestin. J. Neural. Transm. 2014, 121, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Benovic, J.L.; Codina, J.C.; Caron, M.G.; Lefkowitz, R.J. beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef]
- Schmidt, P.; Holsboer, F.; Spengler, D. Beta(2)-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein beta gamma-subunits and the phosphoinositide 3-kinase pathway. Mol. Endocrinol. 2001, 15, 553–564. [Google Scholar] [PubMed] [Green Version]
- Korn, S.H.; Wouters, E.F.; Wesseling, G.; Arends, J.W.; Thunnissen, F.B. Interaction between glucocorticoids and beta2-agonists: Alpha and beta glucocorticoid-receptor mRNA expression in human bronchial epithelial cells. Biochem. Pharmacol. 1998, 56, 1561–1569. [Google Scholar] [CrossRef]
- Leach, S.; Suzuki, K. Adrenergic Signaling in Circadian Control of Immunity. Front. Immunol. 2020, 11, 1235. [Google Scholar] [CrossRef] [PubMed]
- Shimba, A.; Ikuta, K. Glucocorticoids Regulate Circadian Rhythm of Innate and Adaptive Immunity. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Billings, M.E.; Gold, D.; Szpiro, A.; Aaron, C.P.; Jorgensen, N.; Gassett, A.; Leary, P.J.; Kaufman, J.D.; Redline, S.R. The association of ambient air pollution with sleep apnea: The Multi-Ethnic Study of Atherosclerosis. Ann. Am. Thorac. Soc. 2019, 16, 363–370. [Google Scholar] [CrossRef]
- Cantone, L.; Tobaldini, E.; Favero, C.; Albetti, B.; Sacco, R.M.; Torgano, G.; Ferrari, L.; Montano, N.; Bollati, V. Particulate Air Pollution, Clock Gene Methylation, and Stroke: Effects on Stroke Severity and Disability. Int. J. Mol. Sci. 2020, 21, 3090. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, T.S.; Saenen, N.D.; Schenk, J.; Janssen, B.G.; Motta, V.; Tarantini, L.; Cox, B.; Lefebvre, W.; Vanpoucke, C.; Maggioni, C.; et al. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ. Int. 2018, 114, 231–241. [Google Scholar] [CrossRef]
- Palanivel, R.; Vinayachandran, V.; Biswal, S.; Deiuliis, J.A.; Padmanabhan, R.; Park, B.; Gangwar, R.S.; Durieux, J.C.; Cara, E.A.E.; Das, L.; et al. Exposure to Air Pollution Disrupts Circadian Rhythm through Alterations in Chromatin Dynamics. iScience 2020, 23, 101728. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; McQueen, A.; Chen, T.-C.; Wang, J.-C. Regulation of Glucose Homeostasis by Glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar]
- Druzd, D.; Matveeva, O.; Ince, L.; Harrison, U.; He, W.; Schmal, C.; Herzel, H.; Tsang, A.H.; Kawakami, N.; Leliavski, A.; et al. Lymphocyte Circadian Clocks Control Lymph Node Trafficking and Adaptive Immune Responses. Immunity 2017, 46, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Ralph, M.R.; Foster, R.G.; Davis, F.C.; Menaker, M. Transplanted suprachiasmatic nucleus determines circadian period. Science 1990, 247, 975–978. [Google Scholar] [CrossRef] [Green Version]
- De Boer, S.; Van Der Gugten, J. Daily variations in plasma noradrenaline, adrenaline and corticosterone concentrations in rats. Physiol. Behav. 1987, 40, 323–328. [Google Scholar] [CrossRef]
- Jin, X.; Shearman, L.P.; Weaver, D.R.; Zylka, M.J.; De Vries, G.J.; Reppert, S.M. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell 1999, 96, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Kume, K.; Zylka, M.J.; Sriram, S.; Shearman, L.P.; Weaver, D.R.; Jin, X.; Maywood, E.S.; Hastings, M.H.; Reppert, S.M. mCRY1 and mCRY2 Are Essential Components of the Negative Limb of the Circadian Clock Feedback Loop. Cell 1999, 98, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Hobson, J.; McCarley, R.; Wyzinski, P. Sleep cycle oscillation: Reciprocal discharge by two brainstem neuronal groups. Sci. 1975, 189, 55–58. [Google Scholar] [CrossRef] [PubMed]
- González, M.M.; Aston-Jones, G. Circadian regulation of arousal: Role of the noradrenergic locus coeruleus system and light exposure. Sleep 2006, 29, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, S.; Undem, B.J. Vagal Afferent Innervation of the Airways in Health and Disease. Physiol. Rev. 2016, 96, 975–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.; Salazar, R.; Wilson, T.; Lucas, S.; Herbert, G.; Young, T.; Begay, J.; Denson, J.L.; Zychowski, K.; Ashley, R.; et al. Early Gestational Exposure to Inhaled Ozone Impairs Maternal Uterine Artery and Cardiac Function. Toxicol. Sci. 2020, 179, 121–134. [Google Scholar] [CrossRef]
- Aragon, M.J.; Topper, L.; Tyler, C.R.; Sanchez, B.; Zychowski, K.; Young, T.; Herbert, G.; Hall, P.; Erdely, A.; Eye, T.; et al. Serum-borne bioactivity caused by pulmonary multiwalled carbon nanotubes induces neuroinflammation via blood–brain barrier impairment. Proc. Natl. Acad. Sci. USA 2017, 114, E1968–E1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chaudhury, D.; Nectow, A.R.; Friedman, A.K.; Zhang, S.; Juarez, B.; Liu, H.; Pfau, M.L.; Aleyasin, H.; Jiang, C.; et al. α1- and β3-Adrenergic Receptor–Mediated Mesolimbic Homeostatic Plasticity Confers Resilience to Social Stress in Susceptible Mice. Biol. Psychiatry 2019, 85, 226–236. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Type | Affinity for Endogenous Substrate | Tissue Distribution | Cellular Response | References |
|---|---|---|---|---|
| AR-α1 | EPI ≥ NE | Vascular smooth muscle, heart | Vasoconstriction | [28,29,32] |
| AR-α2 | EPI ≥ NE | Presynaptic adrenergic and cholinergic nerve terminals (postsynaptic CNS) | Inhibition of transmitter release (SNS outflow reduction) | [29,32] |
| AR-β1 | EPI = NE | Heart, kidney (glomerular cells) | Increases heart rate, ventricular muscle contraction, increases renin release | [28,30,32,33] |
| AR-β2 | EPI >> NE | Smooth muscle cells (respiratory, vascular, and uterine), respiratory epithelial cells | Increases smooth muscle relaxation, increases glucose in liver, fluid balance, proinflammatory (increases contractility in the heart) | [1,28,32] |
| AR-β3 | NE > EPI | Adipose tissue cells, bladder | Increases lipolysis in adipose tissue and relaxes the bladder muscle | [31,32] |
| GR-α | Endogenous glucocorticoids | All tissue and cell types | Nuclear translocation, activation/inhibition of genes, nongenomic regulation of cellular processes | [25,26] |
| GR-β | No ligand | All tissues, abundant in neutrophils and epithelial cells | Localized in the nucleus, inhibits GR-alpha activity, involved in glucocorticoid resistance | [25] |
| Pollutant Type | Model System | Receptor Subtype | Study Design and Outcome | Reference |
|---|---|---|---|---|
| Ambient PM | Human trial | Endogenous ligands for ARs and GRs | PM exposure increased cortisol, epinephrine, norepinephrine, and changed glucose and lipid metabolites in serum. | [89] |
| Ambient NO2 (Traffic) | Epidemiology | Endogenous GR ligand | NO2 but not PM exposure was associated with increased morning cortisol in plasma. | [90] |
| Ambient pollutants | Epidemiology | Endogenous AR ligand | Ambient pollution was associated with increases in urine catecholamines. | [91] |
| Ambient PM | Dog | αARs | Dogs exposed to ambient PM through tracheal tube had increased blood pressure, and this PM effect was inhibited by αAR antagonists. | [88] |
| Cigarette smoke * | Lung epithelial cell line | β2AR-associated second messengers | Suppression of inflammatory cytokine production through β-arrestin signaling was linked to βARs and inhibition of autophagy through AMPK in cigarette-smoke-condensate-exposed cells. | [92] |
| LPS | Macrophage cell line | β2AR and β-arrestin | β2AR negatively regulated NF-κB by β-arrestin 2, and through stabilizing the NF-κB/IκB-α complex. | [93] |
| Ambient PM | Mice in vivo, and human macrophages | β2AR and its ligand | PM exposure in mice increased circulating catecholamines and macrophage IL-6 release. In human macrophages, β2AR agonists increased—and antagonists decreased—IL-6 production. | [8] |
| Acrolein | Rat | Endogenous ligands for ARs and GRs | Acrolein inhalation increased corticosterone and epinephrine in Wistar and diabetic Goto–Kakizaki rats, which were associated with nasal injury and inflammation. | [94] |
| Ambient PM | Adra2b-transgenic mice | α2AR | Concentrated PM exposure increased blood pressure, and anxiety-like behavior, which was associated with upregulation of inflammatory genes in the brains of Adra2b-transgenic mice, overexpressing α2bAR. | [95] |
| Diesel exhaust | Endothelial cells | βARs | In endothelial cells, diesel exhaust extract increased inflammatory cytokines’ release, and this effect was inhibited by βARs and calcium channel inhibitors in an extract-specific manner. | [3] |
| Ambient PM | Rat microvessels, ex vivo | αARs | Microvessels isolated from PM-exposed rats had inhibited endothelium-dependent arteriolar dilation. αARs inhibited PM effects. | [96] |
| Ambient air pollution | Humans and mice | Endogenous ligands for GRs | Exposure to air pollution was associated with increased plasma cortisol in humans and corticosterone in mice. In mice, PM increased hippocampal inflammation and inhibited GR expression. | [97] |
| Metal mixture | Mouse macrophage cell line | GR activation | GR activity was inhibited by selected metals, as indicated by reporter luciferase assay. | [98] |
| Ambient PM | Rat | GRs | Increased expression of genes regulated by activation of GRs in multiple tissues, including lung. | [99] |
| Model System | Receptor Subtype | Study Results | References |
|---|---|---|---|
| Human | Endogenous ligands for GRs | In a clinical study, ozone exposure increased plasma levels of cortisol, which was associated with increased lipid metabolites | [110] |
| Rat | Endogenous ligands for ARs | Epinephrine level increased in rats immediately after ozone exposure, and this was associated with lung injury inflammation and lymphopenia. | [14,105] |
| Rat | Endogenous ligand manipulation for ARs and GRs | Adrenal demedullation diminished circulating epinephrine, and total adrenalectomy diminished both epinephrine and corticosterone. This was associated with inhibition of ozone-induced lung injury, inflammation, lymphopenia, and lung expression of genes involved in AR and GR signaling, acute-phase response, hypoxia, and inflammation. | [4,113,114] |
| Rat | β2AR and GR agonists, individually or in combination | Pretreatment of rats with β2AR agonists exacerbated ozone-induced lung injury and inflammation. GR agonists, but not β2AR agonists, exacerbated ozone-induced lymphopenia. Combination treatment exacerbated both lymphopenia and lung effects, including gene expression of inflammatory markers and GR-responsive targets, in both sham and adrenalectomized rats. | [4,6,15] |
| Rat | βAR and GR antagonists | βAR antagonists suppressed ozone-induced lung vascular leakage and neutrophilia, while GR antagonists reversed lymphopenia but not lung neutrophilia. The combination of both antagonists inhibited all ozone-induced effects. | [5] |
| Rat | Endogenous ligands of ARs and GRs in brain effects | Depletion of circulating endogenous ligands, epinephrine, and corticosterone by adrenalectomy inhibited ozone-induced changes in gene expression within the brainstem and hypothalamus. This was associated with the reversal of ozone-induced decreases in circulating prolactin, luteinizing hormone, and thyroid-stimulating hormone. | [7] |
| Rat | Endogenous ligands for ARs and GRs | Over a 4-h period of ozone exposure, circulating epinephrine and corticosterone increased. These increases were followed by the depletion of circulating granulocytes, M1 monocytes, B and T lymphocytes, and lung expression of GR-regulated genes. Only small changes occurred in circulating cytokines. | [85] |
| Rat | Endogenous ligands for GRs | Ozone exposure increased corticosterone in lung lavage fluid and inhibited alveolar macrophage cytokine production. The stress-sensitive Fischer 344 strain exhibited greater effects than those of stress-resistant Lewis rats. Inhibiting corticosterone production increased inflammatory cytokine expression in macrophages. | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodge, M.X.; Henriquez, A.R.; Kodavanti, U.P. Adrenergic and Glucocorticoid Receptors in the Pulmonary Health Effects of Air Pollution. Toxics 2021, 9, 132. https://doi.org/10.3390/toxics9060132
Hodge MX, Henriquez AR, Kodavanti UP. Adrenergic and Glucocorticoid Receptors in the Pulmonary Health Effects of Air Pollution. Toxics. 2021; 9(6):132. https://doi.org/10.3390/toxics9060132
Chicago/Turabian StyleHodge, Myles X., Andres R. Henriquez, and Urmila P. Kodavanti. 2021. "Adrenergic and Glucocorticoid Receptors in the Pulmonary Health Effects of Air Pollution" Toxics 9, no. 6: 132. https://doi.org/10.3390/toxics9060132
APA StyleHodge, M. X., Henriquez, A. R., & Kodavanti, U. P. (2021). Adrenergic and Glucocorticoid Receptors in the Pulmonary Health Effects of Air Pollution. Toxics, 9(6), 132. https://doi.org/10.3390/toxics9060132