The Current Status of the Pharmaceutical Potential of Juniperus L. Metabolites



Abstract
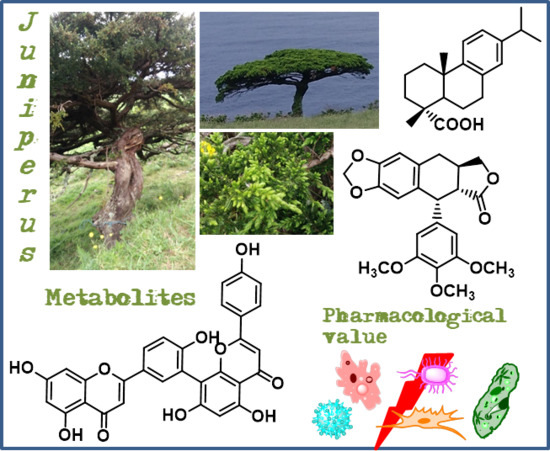
:
1. Introduction
2. Bioactive Secondary Metabolites from Juniperus Species
2.1. Terpenoids
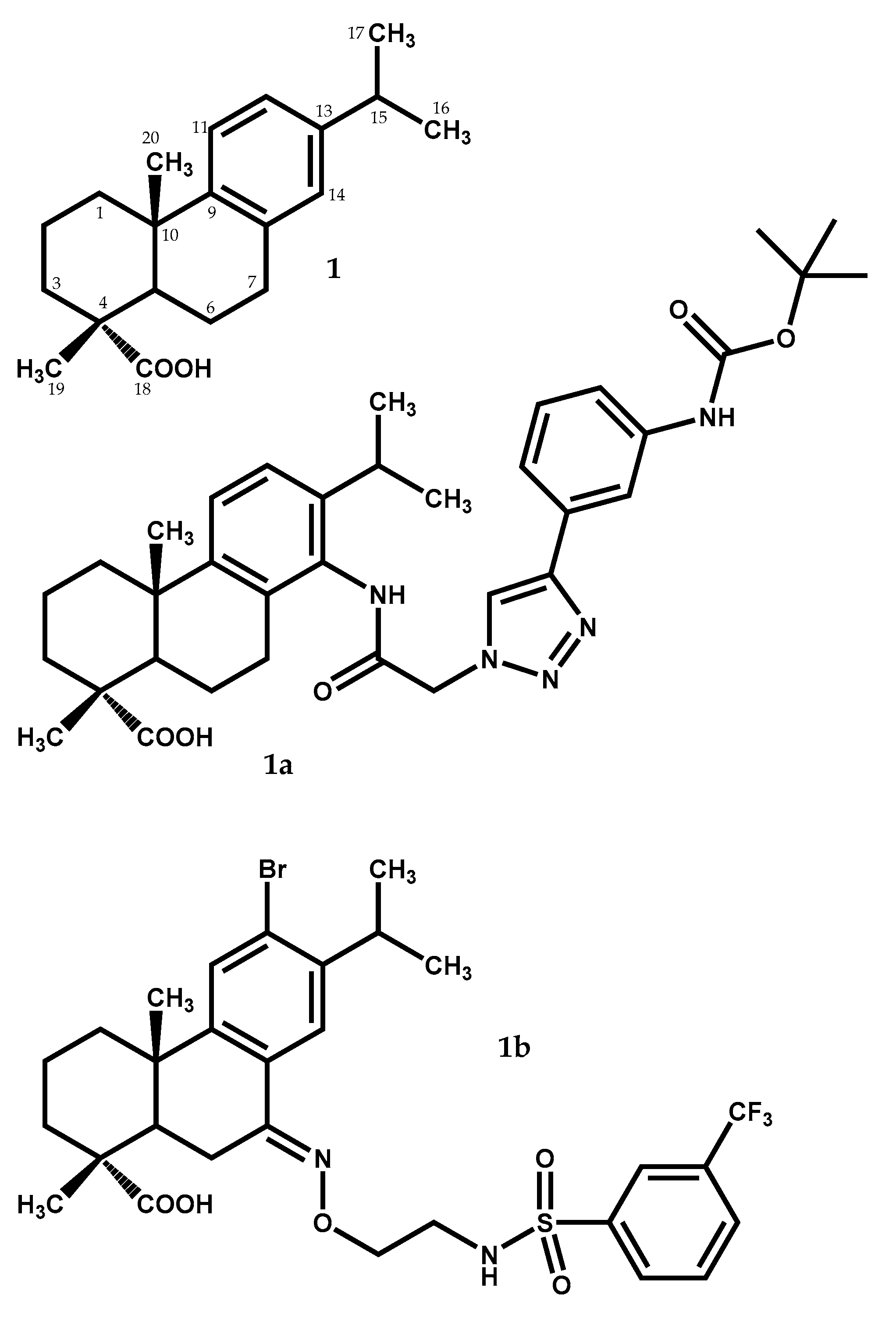
2.1.1. Dehydroabietic Acid
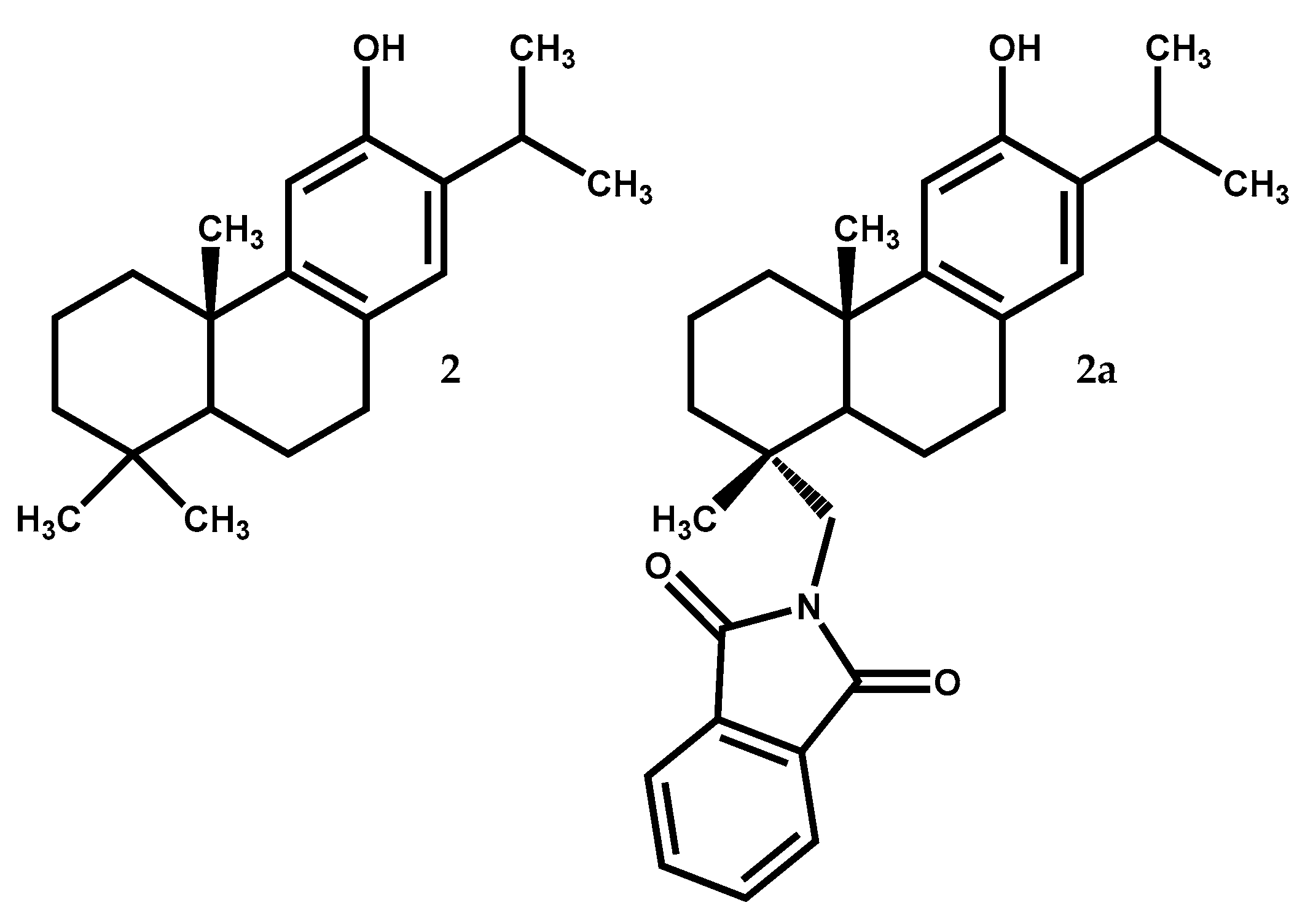
2.1.2. Ferruginol
2.1.3. Hinokiol
2.1.4. Sugiol
2.1.5. Totarol
2.2. Flavonoids
2.2.1. Amentoflavone
2.2.2. Rutin
2.3. Lignans
Deoxypodophyllotoxin
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| 5-Fu | Fluorouracil |
| A375 | Human malignant melanoma |
| A549 | Human lung carcinoma |
| AIF | Apoptosis inducing factor |
| AKR1B10 | Aldo-keto reductase family 1 member B10 |
| AKT/PKB | Protein kinase B |
| AMPK | Adenosine monophosphate -activated protein kinase |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid-β |
| Bax | Bcl-2-associated X |
| BChE | Butyrylcholinesterase |
| BGC-823 | Gastric carcinoma |
| C6 | Rat glial tumor |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| Cdc2 | Cyclin-dependent protein kinase 2 |
| Cdc25C | Cell division cycle 25C protein |
| CGC | Cerebellar granule cells |
| CNS | Central nervous system |
| CoA | Coenzyme A |
| CUS | Chronic unpredictable stress |
| DNA | Deoxyribonucleic acid |
| DNCB | 2,4-Dinitrochlorobenzene |
| DPP-IV | Dipeptidyl peptidase IV |
| DPT | Deoxypodophyllotoxin |
| DPT-HP-β-CD | Mixture of deoxypodophyllotoxin with hydroxypropyl-β-cyclodextrin |
| DU145 | Human prostate cancer |
| DZR | Dexrazoxane |
| EC50 | Half maximal effective concentration |
| EJ138 | Human bladder carcinoma |
| ERK | Extracellular signal-regulated kinase |
| FAD | Flavin adenine dinucleotide |
| GIP | Glucose-dependent insulinotropic polypeptide |
| GLP-1 | Glucagon-like peptide 1 |
| GSH | Glutathione |
| H9c2 | Rat cardiomyoblasts |
| HaCaT | Nontumorigenic human epidermal cells |
| HCT-8 | Human colorectal adenocarcinoma |
| HCT116 | Human colon cancer |
| HeLa | Human cervical carcinoma |
| Hep3B | Human hepatoma |
| HepG2 | Human hepatocellular carcinoma |
| HFF-1 | Human normal fibroblast |
| HL-7702 | Human liver normal |
| HO-8910 | Human ovarian carcinoma |
| HT-29 | Human colon carcinoma |
| HUVEC | Human umbilical vein endothelial cells |
| IC50 | Half maximal inhibitory concentration |
| IgE | Immunoglobulin E |
| IL-4 | Interleukin 4 |
| JeG-3 | Human choriocarcinoma |
| L-02 | Human fetal hepatocyte normal cell line |
| LTP | Long-term potentiation |
| MCF-7/A | Acquired resistant human breast cancer |
| MCF-7/S | Human breast cancer |
| MCF-7 | Human breast cancer |
| MDA-MB-231 | Human breast adenocarcinoma |
| MDCK | Madin-Darby canine kidney cells |
| MGC-803 | Human gastric cancer |
| Mia-PaCa2 | Human pancreatic carcinoma |
| MIC | Minimum inhibitory concentration |
| MMP | Mitochondrial membrane potential |
| MMP-9 | Matrix metallopeptidase 9 |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mTOR | Mammalian target of rapamycin |
| N2A | Mouse neuroblastoma |
| NAD(P)+ | Nicotinamide adenine dinucleotide phosphate |
| NCI-H460 | Human lung carcinoma |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OVCAR-3 | Human ovarian adenocarcinoma |
| PAR | Poly (ADP-ribose) |
| PARP-1 | Poly (ADP-ribose) synthetase 1 |
| PBPK-PD | Physiologically based pharmacokinetic-pharmacodynamic |
| PC-3 | Human prostate cancer |
| PGD2 | Prostaglandin D2 |
| PKB/HO-1 | Protein kinase B/heme oxygenase-1 |
| PKC | Protein kinase C |
| PTEN | Phosphatase and tensin homolog |
| RAW 267.4 | Macrophage normal cell line |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SF126 | Human glioblastoma |
| SGC-7901 | Gastric carcinoma |
| SHG-44 | Human malignant glioma |
| SI | Selective index |
| SK-OV-3 | Ovarian cancer |
| Skp2 | S-phase kinase protein 2 |
| SOD | Superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF | Transforming growth factor |
| TIMP-1 | TIMP metallopeptidase inhibitor 1 |
| TKT | Transketolase |
| TNF-α | Tumor necrosis factor α |
| U-87 MG | Human glioblastoma-astrocytoma |
| VGSC | Voltage-gated Na+ channels |
| WI-38 | Human lung fibroblast normal cells |
References
- Youngs, R.L.; Hamza, M.F. Wood: History of use. In Reference Module in Materials Science and Materials Engineering, 1st ed.; Hashmi, S., Ed.; Elsevier Inc.: Oxford, UK, 2016; pp. 1–7. ISBN 978-0-12-803581-8. [Google Scholar]
- Lukešová, H.; Palau, A.S.; Holst, B. Identifying plant fibre textiles from Norwegian Merovingian period and Viking age graves: The late iron age collection of the University Museum of Bergen. J. Archaeol. Sci. Rep. 2017, 13, 281–285. [Google Scholar] [CrossRef]
- Prinsloo, G.; Nogemane, N.; Street, R. The use of plants containing genotoxic carcinogens as foods and medicine. Food Chem. Toxicol. 2018, 116, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Falzon, C.C.; Balabanova, A. Phytotherapy: An introduction to herbal medicine. Prim. Care Clin. Off. Pract. 2017, 44, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Ghosh, R.C.; Kundu, A.; Mandal, S.C. Herb and drug interaction. In Natural Products and Drug Discovery, 1st ed.; Mandal, S.C., Mandal, V., Konishi, T., Eds.; Elsevier Inc.: Oxford, UK, 2018; pp. 467–490. ISBN 978-0-08-102081-4. [Google Scholar]
- Rangel, M.L.; Guerrero-Analco, J.A.; Monribot-Villanueva, J.L.; Kiel-Martínez, A.L.; Avendaño-Reyes, S.; Abad, J.P.D.; Bonilla-Landa, I.; Dávalos-Sotelo, R.; Olivares-Romero, J.L.; Angeles, G. Anatomical and chemical characteristics of leaves and branches of Juniperus deppeana var. deppeana (Cupressaceae): A potential source of raw materials for the perfume and sweet candies industries. Ind. Crops Prod. 2018, 113, 50–54. [Google Scholar] [CrossRef]
- The Plant List. Available online: http://www.theplantlist.org/1.1/browse/G/Cupressaceae/Juniperus (accessed on 28 May 2018).
- Khan, M.; Khan, A.; Rehman, N.; Gilani, A.H. Pharmacological explanation for the medicinal use of Juniperus excelsa in hyperactive gastrointestinal and respiratory disorders. J. Nat. Med. 2012, 66, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Orhan, N.; Aslan, M.; Pekcan, M.; Orhan, D.D.; Bedir, E.; Ergun, F. Identification of hypoglycaemic compounds from berries of Juniperus oxycedrus subsp. oxycedrus through bioactivity guided isolation technique. J. Ethnopharmacol. 2012, 139, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Flores-Vallejo, R.C.; Cardoso-Taketa, A.; Villarreal, M.L. Antibacterial activities of medicinal plants used in Mexican traditional medicine. J. Ethnopharmacol. 2017, 208, 264–329. [Google Scholar] [CrossRef] [PubMed]
- Bais, S.; Gill, N.S.; Rana, N.; Shandil, S. A Phytopharmacological review on a medicinal plant: Juniperus communis. Int. Sch. Res. Not. 2014, 2014, 634723. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Silva, A.M.S. The chemical composition of the Juniperus genus (1970–2004). In Recent Progress in Medicinal Plants; Govil, J.N., Singh, V.K., Bhardwaj, R., Eds.; Studium Press LLC: Houston, TX, USA, 2006; Volume 16, pp. 401–522. ISBN 0-9761849–8–2. [Google Scholar]
- Seca, A.M.L.; Pinto, D.C.G.A.; Silva, A.M.S. The current status of bioactive metabolites from the genus Juniperus. In Bioactive Phytochemicals: Perspectives for Modern Medicine; Gupta, V.K., Ed.; M/S Daya Publishing House: New Delhi, India, 2015; Volume 3, pp. 365–407. ISBN 9789351246749. [Google Scholar]
- Al-Snafi, A.E. Medical importance of Juniperus communis—A review. Indo Am. J. Pharm. Sci. 2018, 5, 1779–1792. [Google Scholar] [CrossRef]
- Al-Snafi, A.E. Pharmacological and therapeutic effects of Juniperus oxycedrus—A review. Indo Am. J. Pharm. Sci. 2018, 5, 2198–2205. [Google Scholar] [CrossRef]
- Jung, H.J.; Min, B.-S.; Jung, H.A.; Choi, J.S. Sesquiterpenoids from the heartwood of Juniperus chinensis. Nat. Prod. Sci. 2017, 23, 208–212. [Google Scholar] [CrossRef]
- Lee, S.; Park, N.-J.; Bong, S.-K.; Jegal, J.; Park, S.-A.; Kim, S.-N.; Yang, M.H. Ameliorative effects of Juniperus rigida fruit on oxazolone- and 2,4-dinitrochlorobenzene-induced atopic dermatitis in mice. J. Ethnopharmacol. 2018, 214, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Groshi, A.A.; Evans, A.R.; Ismail, F.M.D.; Nahar, L.; Sarker, S.D. Cytotoxicity of Libyan Juniperus phoenicea against human cancer cell lines A549, EJ138, HepG2 and MCF7. Pharm. Sci. 2018, 24, 3–7. [Google Scholar] [CrossRef]
- Venditti, A.; Maggi, F.; Quassinti, L.; Bramucci, M.; Lupidi, G.; Ornano, L.; Ballero, M.; Sanna, C.; Bruno, M.; Rosselli, S.; et al. Bioactive constituents of Juniperus turbinata Gussone from La Maddalena Archipelago. Chem. Biodivers. 2018, e1800148. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Cavadas, C.; Cavaleiro, C.; Salgueiro, L.; do Céu Sousa, M. In vitro susceptibility of Trypanosoma brucei brucei to selected essential oils and their major components. Exp. Parasitol. 2018, 190, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yao, L. The anxiolytic effect of Juniperus virginiana L. essential oil and determination of its active constituents. Physiol. Behav. 2018, 189, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Inci, H.; Ozdemir, G.; Sengul, A.Y.; Sogut, B.; Nursoy, H.; Sengul, T. Using juniper berry (Juniperus communis) as a supplement in Japanese quail diets. Rev. Bras. Zootec. 2016, 45, 230–235. [Google Scholar] [CrossRef]
- Brischke, C.; Hesse, C.; Meyer-Veltrup, L.; Humar, M. Studies on the material resistance and moisture dynamics of Common juniper, English yew, Black cherry, and Rowan. Wood Mater. Sci. Eng. 2018, 13, 222–230. [Google Scholar] [CrossRef]
- Ateş, S.; Gür, M.; Özkan, O.E.; Akça, M.; Olgun, Ç.; Güder, A. Chemical contents and antifungal activity of some durable wood extractives vs. Pleurotus ostreatus. Bioresources 2015, 10, 2433–2443. [Google Scholar]
- Koruk, S.T.; Ozyilkan, E.; Kava, P.; Colak, D.; Donderici, O.; Cesaretli, Y. Juniper tar poisoning. Clin. Toxicol. 2005, 43, 47–49. [Google Scholar] [CrossRef]
- González, M.A. Aromatic abietane diterpenoids: Their biological activity and synthesis. Nat. Prod. Rep. 2015, 32, 684–704. [Google Scholar] [CrossRef] [PubMed]
- Agudelo-Gómez, L.S.; Betancur-Galvis, L.A.; González, M.A. Anti HHV-1 and HHV-2 activity in vitro of abietic and dehydroabietic acid derivatives. Pharmacologyonline 2012, 1, 36–42. [Google Scholar]
- Zaidi, S.F.H.; Awale, S.; Kalauni, S.K.; Tezuka, Y.; Esumi, H.; Kadota, S. Diterpenes from “Pini Resina” and their preferential cytotoxic activity under nutrient-deprived condition. Planta Med. 2006, 72, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Tokuda, H.; Ezaki, Y. Cancer chemopreventive activity of “rosin” constituents of Pinus spez. and their derivatives in two-stage mouse skin carcinogenesis test. Phytomedicine 2008, 15, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, B.; Astudillo, L.; Rodríguez, J.A.; Yáñez, T.; Theoduloz, C.; Schmeda-Hirschmann, G. Gastroprotective and cytotoxic effect of dehydroabietic acid derivatives. Pharmacol. Res. 2005, 52, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Fallarero, A.; Skogman, M.; Kujala, J.; Rajaratnam, M.; Moreira, V.M.; Yli-Kauhaluoma, J.; Vuorela, P. (+)-Dehydroabietic Acid, an Abietane-Type Diterpene, Inhibits Staphylococcus aureus Biofilms in vitro. Int. J. Mol. Sci. 2013, 14, 12054–12072. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Yang, K.-S. Inhibition of nitric oxide production in RAW 264.7 macrophages by diterpenoids from Phellinus pini. Arch. Pharm. Res. 2011, 34, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Luo, Z.; Zhang, G.; Cao, D.; Li, D.; Ruan, H.; Ruan, B.H.; Su, L.; Xu, H. Click chemistry-based synthesis and anticancer activity evaluation of novel C-14 1,2,3-triazole dehydroabietic acid hybrids. Eur. J. Med. Chem. 2017, 138, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-M.; Yao, Y.; Yang, T.; Wang, X.-Y.; Zhu, Z.-Y.; Xu, W.-T.; Lin, H.-X.; Gao, Z.-B.; Zhou, H.; Yang, C.-G.; et al. The synthesis and antistaphylococcal activity of N-sulfonaminoethyloxime derivatives of dehydroabietic acid. Bioorg. Med. Chem. Lett. 2018, 28, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-W.; Shim, D.-W.; Shin, W.-Y.; Kim, M.-K.; Shim, E.-J.; Sun, X.; Koppula, S.; Kim, T.-J.; Kang, T.-B.; Lee, K.-H. Juniperus rigida Sieb. extract inhibits inflammatory responses via attenuation of TRIF-dependent signaling and inflammasome activation. J. Ethnopharmacol. 2016, 190, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Samoylenko, V.; Dunbar, D.C.; Gafur, M.A.; Khan, S.I.; Ross, S.A.; Mossa, J.S.; El-Feraly, F.S.; Tekwani, B.L.; Bosselaers, J.; Muhammad, I. Antiparasitic, nematicidal and antifouling constituents from Juniperus berries. Phytother. Res. 2008, 22, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Becerra, J.; Flores, C.; Mena, J.; Aqueveque, P.; Alarcón, J.; Bittner, M.; Hernández, V.; Hoeneisen, M.; Ruiz, E.; Silva, M. Antifungal and antibacterial activity of diterpenes isolated from wood extractables of Chilean Podocarpaceae. Bol. Soc. Chil. Quim. 2002, 47, 151–157. [Google Scholar] [CrossRef]
- Mossa, J.S.; El-Feraly, F.S.; Muhammad, I. Antimycobacterial constituents from Juniperus procera, Ferula communis and Plumbago zeylanica and their in vitro synergistic activity with isonicotinic acid hydrazide. Phytother. Res. 2004, 18, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.J.; Williamson, E.M.; Wareham, N.; Kaatz, G.W.; Gibbons, S. Antibacterials and modulators of bacterial resistance from the immature cones of Chamaecyparis lawsoniana. Phytochemistry 2007, 68, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.J.; Wareham, N.; Zloh, M.; Gibbons, S. 2b-Acetoxyferruginol—A new antibacterial abietane diterpene from the bark of Prumnopitys andina. Phytochem. Lett. 2008, 1, 49–53. [Google Scholar] [CrossRef]
- Ryu, Y.B.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, J.-Y.; Kim, D.; Naguyen, T.T.H.; Park, S.-J.; Chang, J.S.; Park, K.H.; et al. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorg. Med. Chem. 2010, 18, 7940–7947. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, C.; Chen, H.; Huang, M.; Ma, X.; Deng, S.; Huang, Y.; Wen, Y.; Yang, X.; Song, P. In vitro and in vivo antitumor effects of the diterpene-enriched extract from Taxodium ascendens through the mitochondrial-dependent apoptosis pathway. Biomed. Pharmacother. 2017, 96, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid β-protein dimers isolated directly from Alzheimer brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Zolezzi, J.M.; Lindsay, C.B.; Serrano, F.G.; Ureta, R.C.; Theoduloz, C.; Schmeda-Hirschmann, G.; Inestrosa, N.C. Neuroprotective effects of ferruginol, jatrophone, and junicedric acid against amyloid-β injury in hippocampal neurons. J. Alzheimer’s Dis. 2018, 63, 705–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-W.; Hu, X.-D.; Zhang, H.-M.; Xin, W.-J.; Li, M.-T.; Zhang, T.; Zhou, L.-J.; Liu, X.-G. Roles of CaMKII, PKA, and PKC in the induction and maintenance of LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J. Neurophysiol. 2004, 91, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Roa-Linares, V.C.; Brand, Y.M.; Agudelo-Gomez, L.S.; Tangarife-Castaño, V.; Betancur-Galvis, L.A.; Gallego-Gomez, J.C.; González, M.A. Anti-herpetic and anti-dengue activity of abietane ferruginol analogues synthesized from (+)-dehydroabietylamine. Eur. J. Med. Chem. 2016, 108, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.-Y.; Zeng, H.-W.; Pei, Y.-H.; Li, L.; Ye, J.; Pan, Y.-X.; Zhang, J.-G.; Yuan, X.; Zhang, W.-D. The anti-inflammatory activities of an extract and compounds isolated from Platycladus orientalis (Linnaeus) Franco in vitro and ex vivo. J. Ethnopharmacol. 2012, 141, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Li, Y.-C.; You, B.-J.; Chang, W.-T.; Chao, L.K.; Lo, L.-C.; Wang, S.-Y.; Huang, G.-J.; Kuo, Y.-H. Diterpenoids with anti-inflammatory activity from the wood of Cunninghamia konishii. Molecules 2013, 18, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Marques, C.; Simões, M.F.; Valdeira, M.L.; Rodríguez, B. Terpenoids and phenolics from Plectranthus strigosus. Nat. Prod. Res. 2008, 22, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Alqasoumi, S.I.; Abdel-Kader, M.S. Terpenoids from Juniperus procera with hepatoprotective activity. Pak. J. Pharm. Sci. 2012, 25, 315–322. [Google Scholar] [PubMed]
- Wang, W.-S.; Li, E.-W.; Jia, Z.-J. Terpenes from Juniperus przewalskii and their antitumor activities. Pharmazie 2002, 57, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Yang, C.-T.; Gong, C.-L.; Chen, Y.-H.; Chen, Y.-W.; Wu, K.-C.; Cheng, T.-H.; Kuo, Y.-H.; Chen, Y.-F.; Leung, Y.-M. Inhibition of voltage-gated Na+ channels by hinokiol in neuronal cells. Pharmacol. Rep. 2015, 67, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Silva, A.M.S. The chemical composition of hexane extract from bark of Juniperus brevifolia. Nat. Prod. Res. 2008, 22, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Silva, A.M.S.; Bazzocchi, I.L.; Jimenez, I.A. Diterpene constituents of leaves from Juniperus brevifolia. Phytochemistry 2008, 69, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, V.K.; Sharma, A.; Kang, S.C.; Baek, K.-H. Antioxidant, lipid peroxidation inhibition and free radical scavenging efficacy of a diterpenoid compound sugiol isolated from Metasequoia glyptostroboides. Asian Pac. J. Trop. Med. 2014, 7, 9–15. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Kang, S.C. A diterpenoid sugiol from Metasequoia glyptostroboides with α-glucosidase and tyrosinase inhibitory potential. Bangladesh J. Pharmacol. 2014, 9, 312–316. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Kim, N.-H.; Kim, K.; Kang, S.C. Antiviral potential of a diterpenoid compound sugiol from Metasequoia glyptostroboides. Pak. J. Pharm. Sci. 2016, 29, 1077–1080. [Google Scholar] [PubMed]
- Jung, S.-N.; Shin, D.-S.; Kim, H.-N.; Jeon, Y.J.; Yun, J.; Lee, Y.-J.; Kang, J.S.; Han, D.C.; Kwon, B.-M. Sugiol inhibits STAT3 activity via regulation of transketolase and ROS-mediated ERK activation in DU145 prostate carcinoma cells. Biochem. Pharmacol. 2015, 97, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Ma, D.; Lee, W.-N.; Xiao, J.; Zhao, Y.; Go, V.L.; Wang, Q.; Yen, Y.; Recker, R.; et al. Inhibition of transketolase by oxythiamine altered dynamics of protein signals in pancreatic cancer cells. Exp. Hematol. Oncol. 2013, 2, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Zhang, X.; Zhang, H.; Shang, H.; Bao, J.; Wang, H.; Li, Z. Sugiol (12-hydroxyabieta-8,11,13-trien-7-one) targets human pancreatic carcinoma cells (Mia-PaCa2) by inducing apoptosis, G2/M cell cycle arrest, ROS production and inhibition of cancer cell migration. J. BUON 2018, 23, 205–210. [Google Scholar] [PubMed]
- Cox, R.E.; Yamamoto, S.; Otto, A.; Simoneit, B.R.T. Oxygenated di- and tricyclic diterpenoids of southern hemisphere conifers. Biochem. Syst. Ecol. 2007, 35, 342–362. [Google Scholar] [CrossRef]
- Jaiswal, R.; Beuria, T.K.; Mohan, R.; Mahajan, S.K.; Panda, D. Totarol inhibits bacterial cytokinesis by perturbing the assembly dynamics of FtsZ. Biochemistry 2007, 46, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.B.; O’Brien, T.E.; Moore, J.T.; Anderson, D.E.; Foss, M.H.; Weibel, D.B.; Ames, J.B.; Shaw, J.T. The synthesis and antimicrobial activity of heterocyclic derivatives of totarol. ACS Med. Chem. Lett. 2012, 3, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Foss, M.H.; Eun, Y.-J.; Grove, C.I.; Pauw, D.A.; Sorto, N.A.; Rensvold, J.W.; Pagliarini, D.J.; Shaw, J.T.; Weibel, D.B. Inhibitors of bacterial tubulin target bacterial membranes in vivo. Med. Chem. Commun. 2013, 4, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarkson, C.; Musonda, C.C.; Chibale, K.; Campbell, W.E.; Smith, P. Synthesis of totarol amino alcohol derivatives and their antiplasmodial activity and cytotoxicity. Bioorg. Med. Chem. 2003, 11, 4417–4422. [Google Scholar] [CrossRef]
- Tacon, C.; Guantai, E.M.; Smith, P.J.; Chibale, K. Synthesis, biological evaluation and mechanistic studies of totarol amino alcohol derivatives as potential antimalarial agents. Bioorg. Med. Chem. 2012, 20, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.J.; Kaatz, G.W.; Seo, S.M.; Wareham, N.; Williamson, E.M.; Gibbons, S. The phenolic diterpene totarol inhibits multidrug efflux pump activity in Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 4480–4483. [Google Scholar] [CrossRef] [PubMed]
- Gordien, A.Y.; Gray, A.I.; Franzblau, S.G.; Seidel, V. Antimycobacterial terpenoids from Juniperus communis L. (Cuppressaceae). J. Ethnopharmacol. 2009, 126, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.B.; Furneaux, R.H.; Gainsford, G.J.; Murphy, M.P. The synthesis and antibacterial activity of totarol derivatives. Part 3: Modification of ring-B. Bioorg. Med. Chem. 2000, 8, 1663–1675. [Google Scholar] [CrossRef]
- Reddy, P.J.; Ray, S.; Sathe, G.J.; Gajbhiye, A.; Prasad, T.S.K.; Rapole, S.; Panda, D.; Srivastava, S. A comprehensive proteomic analysis of totarol induced alterations in Bacillus subtilis by multipronged quantitative proteomics. J. Proteom. 2015, 114, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, X.; Chang, S.; Wang, Y.; Xu, Y.; Ran, S.; Huang, Z.; Li, P.; Li, J.; Zhang, L.; et al. Totarol prevents neuronal injury in vitro and ameliorates brain ischemic stroke: Potential roles of Akt activation and HO-1 induction. Toxicol. Appl. Pharmacol. 2015, 289, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yan, H.; Zhang, L.; Shan, M.; Chen, P.; Ding, A.; Li, S.F.Y. A review on the phytochemistry, pharmacology, and pharmacokinetics of amentoflavone, a naturally-occurring biflavonoid. Molecules 2017, 22, 299. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-W.; Na, Y.; Park, N.-H.; Kim, H.-S.; Ahn, S.M.; Kim, J.W.; Kim, H.-K.; Jang, Y.P. Amentoflavone inhibits UVB-induced matrix metalloproteinase-1 expression through the modulation of AP-1 components in normal human fibroblasts. Appl. Biochem. Biotechnol. 2012, 166, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.-S.; Lee, J.; Jin, H.-G.; Woo, E.-R.; Lee, D.G. Amentoflavone stimulates mitochondrial dysfunction and induces apoptotic cell death in Candida albicans. Mycopathologia 2012, 173, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Coulerie, P.; Nour, M.; Maciuk, A.; Eydoux, C.; Guillemot, J.-C.; Lebouvier, N.; Hnawia, E.; Leblanc, K.; Lewin, G.; Canard, B.; et al. Structure-activity relationship study of biflavonoids on the Dengue virus polymerase DENV-NS5 RdRp. Planta Med. 2013, 79, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Han, W.; Mai, W.; Han, L.; Chen, D. Amentoflavone protects against hydroxyl radical-induced DNA damage via antioxidant mechanism. Turk. J. Biochem. 2014, 39, 30–36. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Almowallad, F.M.; Esmat, A.; Shehata, I.A.; Abdel-Sattar, E.A. Anti-inflammatory activity of flavonoids from Chrozophora tinctoria. Phytochem. Lett. 2015, 13, 74–80. [Google Scholar] [CrossRef]
- Laishram, S.; Sheikh, Y.; Moirangthem, D.S.; Deb, L.; Pal, B.C.; Talukdar, N.C.; Borah, J.C. Anti-diabetic molecules from Cycas pectinata Griff. traditionally used by the Maiba-Maibi. Phytomedicine 2015, 22, 23–26. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Li, Z.; Dong, Y.; Ren, J.; Huo, J. Amentoflavone protects against psoriasis-like skin lesion through suppression of NF-κB-mediated inflammation and keratinocyte proliferation. Mol. Cell. Biochem. 2016, 413, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.I.; Benítez, W.V.; Colín, A.; Bye, R.; Ríos-Gómez, R.; Calzada, F. Evaluation of the diuretic activity in two Mexican medicinal species: Selaginella nothohybrida and S. lepidophylla and its effects with ciclooxigenases inhibitors. J. Ethnopharmacol. 2015, 163, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Chen, W.-L.; Liu, Y.-C. Amentoflavone induces anti-angiogenic and anti-metastatic effects through suppression of NF-κB activation in MCF-7 cells. Anticancer Res. 2015, 35, 6685–6694. [Google Scholar] [PubMed]
- Ndongo, J.T.; Issa, M.E.; Messi, A.N.; Mbing, J.N.; Cuendet, M.; Pegnyemb, D.E.; Bochet, C.G. Cytotoxic flavonoids and other constituents from the stem bark of Ochna schweinfurthiana. Nat. Prod. Res. 2015, 29, 1684–1687. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.J.; Hwang, L.; Lee, M.; Lee, K.Y.; Ahn, M.-J.; Sung, S.H. Neuroprotective biflavonoids of Chamaecyparis obtusa leaves against glutamate-induced oxidative stress in HT22 hippocampal cells. Food Chem. Toxicol. 2014, 64, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Xu, Z.; Liu, Y.; Xu, L.; Huang, H.; Zhang, J.; Cui, L.; Zhou, C.; Xu, D. Amentoflavone enhances osteogenesis of human mesenchymal stem cells through JNK and p38 MAPK pathways. J. Nat. Med. 2016, 70, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-K.; Liu, C.-X.; Zhai, Y.-Y.; Li, L.-L.; Wang, X.-L.; Feng, W.-S. Protection effect of amentoflavone in Selaginella tamariscina against TNF-α-induced vascular injure of endothelial cells. Acta Pharm. Sin. 2013, 48, 1503–1509. [Google Scholar]
- Garza, L.A.; Liu, Y.; Yang, Z.; Alagesan, B.; Lawson, J.A.; Norberg, S.M.; Loy, D.E.; Zhao, T.; Blatt, H.B.; Stanton, D.C.; et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Sci. Transl. Med. 2012, 4, 126ra34. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.; Tong, H.H.Y.; Ng, K.H.; Lao, C.K.; Chong, C.I.; Chao, C.M. In silico prediction of prostaglandin D2 synthase inhibitors from herbal constituents for the treatment of hair loss. J. Ethnopharmacol. 2015, 175, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Brodie, A.; Sabnis, G.; Jelovac, D. Aromatase and breast cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Tascioglu, A.; Ozcan, S.; Akdemir, A.; Orhan, H.G. In vitro and in silico evaluation of aromatase inhibitory activity of apigenin and amentoflavone; dual benefit of St. John’s Wort in postmenopausal women. Toxicol. Lett. 2016, 258, S125. [Google Scholar] [CrossRef]
- Bais, S.; Abrol, N.; Prashar, Y.; Kumari, R. Modulatory effect of standardised amentoflavone isolated from Juniperus communis L. against Freund’s adjuvant induced arthritis in rats (histopathological and X Ray analysis). Biomed. Pharmacother. 2017, 86, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Qin, L.; Huang, F.; Wang, X.; Yang, L.; Shi, H.; Wu, H.; Zhang, B.; Chen, Z.; Wu, X. Amentoflavone protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through PI3K/Akt and ERK signaling pathways. Toxicol. Appl. Pharmacol. 2017, 319, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yue, Q.; He, S. Amentoflavone suppresses tumor growth in ovarian cancer by modulating Skp2. Life Sci. 2017, 189, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-J.; Lee, E.H.; Lee, C.G.; Rhee, K.-J.; Jung, W.-S.; Choi, Y.; Pan, C.-H.; Kang, K. AKR1B10-inhibitory Selaginella tamariscina extract and amentoflavone decrease the growth of A549 human lung cancer cells in vitro and in vivo. J. Ethnopharmacol. 2017, 202, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.-S.; Kundu, J.; Kundu, J.K.; Surh, Y.-J. Targeting Nrf2-Keap1 signaling for chemoprevention of skin carcinogenesis with bioactive phytochemicals. Toxicol. Lett. 2014, 229, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wahyudi, L.D.; Jeong, J.; Yang, H.; Kim, J.-H. Amentoflavone-induced oxidative stress activates NF-E2-related factor 2 via the p38 MAP kinase-AKT pathway in human keratinocytes. Int. J. Biochem. Cell Biol. 2018, 99, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-L.; Lee, W.-J.; Chen, C.-C.; Lu, C.H.; Chen, C.-H.; Chou, Y.-C.; Lee, I.-T.; Sheu, W.H.-H.; Wu, J.-Y.; Yang, C.-F.; et al. Pharmacogenetics of dipeptidyl peptidase 4 inhibitors in a Taiwanese population with type 2 diabetes. Oncotarget 2017, 8, 18050–18058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beidokhti, M.N.; Lobbens, E.S.; Rasoavaivo, P.; Staerk, D.; Jäger, A.K. Investigation of medicinal plants from Madagascar against DPP-IV linked to type 2 diabetes. S. Afr. J. Bot. 2018, 115, 113–119. [Google Scholar] [CrossRef]
- Yaglioglu, A.S.; Eser, F. Screening of some Juniperus extracts for the phenolic compounds and their antiproliferative activities. S. Afr. J. Bot. 2017, 113, 29–33. [Google Scholar] [CrossRef]
- Ganeshpurkar, A.; Saluja, A.K. The pharmacological potential of rutin. Saudi Pharm. J. 2017, 25, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Gullón, B.; Lú-Chau, T.A.; Moreira, M.T.; Lema, J.M.; Eibes, G. Rutin: A review on extraction, identification and purification methods, biological activities and approaches to enhance its bioavailability. Trends Food Sci. Technol. 2017, 67, 220–235. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Sun, B.; Tong, Q.; Ren, L. Rutin protects against pirarubicin-induced cardiotoxicity through TGF-β1-p38 MAPK signaling pathway. Evid. Based Complement. Altern. Med. 2017, 2017, 1759385. [Google Scholar] [CrossRef] [PubMed]
- Parashar, A.; Mehta, V.; Udayabanu, M. Rutin alleviates chronic unpredictable stress-induced behavioral alterations and hippocampal damage in mice. Neurosci. Lett. 2017, 656, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, S.; Reddy, B.R.; Sudhakar, S.R.; Saxena, S.; Das, T.; Meghah, V.; Swamy, C.V.B.; Kumar, A.; Idris, M.M. Chronic unpredictable stress (CUS)-induced anxiety and related mood disorders in a zebrafish model: Altered brain proteome profile implicates mitochondrial dysfunction. PLoS ONE 2013, 8, e63302. [Google Scholar] [CrossRef] [PubMed]
- Renouard, S.; Lopez, T.; Hendrawati, O.; Dupre, P.; Doussot, J.; Falguieres, A.; Ferroud, C.; Hagege, D.; Lamblin, F.; Laine, E.; et al. Podophyllotoxin and deoxypodophyllotoxin in Juniperus bermudiana and 12 other Juniperus species: Optimization of extraction, method validation, and quantification. J. Agric. Food Chem. 2011, 59, 8101–8107. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Chen, Q.; Kasimu, R.; Aisa, H.A. Isolation of deoxypodophyllotoxin and podophyllotoxin from Juniperus sabina by high speed counter current chromatography. Afinidad 2016, 73, 236–239. [Google Scholar]
- Muto, N.; Tomokuni, T.; Haramoto, M.; Tatemoto, H.; Nakanishi, T.; Inatomi, Y.; Murata, H.; Inada, A. Isolation of apoptosis and differentiation inducing substances toward human promyelocytic leukemia HL-60 cells from leaves of Juniperus taxifolia. Biosci. Biotechnol. Biochem. 2008, 72, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wu, M.; Miao, J.; Duan, H.; Zhang, S.; Chen, M.; Sun, L.; Wang, Y.; Zhang, X.; Zhu, X.; et al. Deoxypodophyllotoxin exerts both anti-angiogenic and vascular disrupting effects. Int. J. Biochem. Cell Biol. 2013, 45, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Jiang, Z.; Duan, H.; Sun, L.; Zhang, S.; Chen, M.; Wang, Y.; Gao, Q.; Song, Y.; Zhu, X.; et al. Deoxypodophyllotoxin triggers necroptosis in human non-small cell lung cancer NCI-H460 cells. Biomed. Pharmacother. 2013, 67, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-R.; Xu, Y.; Jiang, Z.-Z.; Guerram, M.; Wang, B.; Zhu, X.; Zhang, L.-Y. Deoxypodophyllotoxin induces G2/M cell cycle arrest and apoptosis in SGC-7901 cells and inhibits tumor growth in vivo. Molecules 2015, 20, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, B.; Guerram, M.; Sun, L.; Shi, W.; Tian, C.; Zhu, X.; Jiang, Z.; Zhang, L. Deoxypodophyllotoxin suppresses tumor vasculature in HUVECs by promoting cytoskeleton remodeling through LKB1-AMPK dependent Rho A activation. Oncotarget 2015, 6, 29497–29512. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Moon, T.C.; Quan, Z.; Lee, E.; Kim, Y.K.; Yang, J.H.; Suh, S.-J.; Jeong, T.C.; Lee, S.H.; Kim, C.-H.; et al. The naturally occurring flavolignan, deoxypodophyllotoxin, inhibits lipopolysaccharide-induced iNOS expression through the NF-κB activation in RAW264.7 macrophage cells. Biol. Pharm. Bull. 2008, 31, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Guerram, M.; Jiang, Z.-Z.; Sun, L.; Zhu, X.; Zhang, L.-Y. Antineoplastic effects of deoxypodophyllotoxin, a potent cytotoxic agent of plant origin, on glioblastoma U-87 MG and SF126 cells. Pharmacol. Rep. 2015, 67, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Wang, G.; Cai, Q.; Zheng, X.; Zhang, J.; Chen, Q.; Wu, B.; Zhu, X.; Hao, H.; Zhou, F. A promising microtubule inhibitor deoxypodophyllotoxin exhibits better efficacy to multidrug-resistant breast cancer than paclitaxel via avoiding efflux transport. Drug Metab. Dispos. 2018, 46, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Khaled, M.; Belaaloui, G.; Jiang, Z.-Z.; Zhu, X.; Zhang, L.-Y. Antitumor effect of deoxypodophyllotoxin on human breast cancer xenograft transplanted in BALB/c nude mice model. J. Infect. Chemother. 2016, 22, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Pinto, D.C.G.A. Plant secondary metabolites as anticancer agents: Successes in clinical trials and therapeutic application. Int. J. Mol. Sci. 2018, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhou, Q.; Wu, W.-R.; Duan, Y.-X.; Gao, Z.-Y.; Li, Y.-W.; Lu, Q. Anticancer effect of deoxypodophyllotoxin induces apoptosis of human prostate cancer cells. Oncol. Lett. 2016, 12, 2918–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lu, B.; Feng, C.; Wang, C.; Wang, Y.; Luo, T.; Feng, J.; Jia, H.; Chi, G.; Luo, Y.; et al. Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett. 2016, 371, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, Y.; Lu, G.; Xie, S.; Ma, Z.; Chen, Z.; Shen, H.-M.; Xia, D. Importance of ROS-mediated autophagy in determining apoptotic cell death induced by physapubescin B. Redox Biol. 2017, 12, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-H.; Son, K.-M.; Kim, K.-Y.; Yu, S.-N.; Park, S.-G.; Kim, Y.-W.; Nam, H.-W.; Suh, J.-T.; Ji, J.-H.; Ahn, S.-C. Deoxypodophyllotoxin induces cytoprotective autophagy against apoptosis via inhibition of PI3K/AKT/mTOR pathway in osteosarcoma U2OS cells. Pharmacol. Rep. 2017, 69, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, K.; Liu, F.; Li, Y.; Zhong, Z.; Hong, S.; Liu, X.; Liu, L. Predicting antitumor effect of deoxypodophyllotoxin in NCI-H460 tumor-bearing mice on the basis of in vitro pharmacodynamics and a physiologically based pharmacokinetic-pharmacodynamic model. Drug Metab. Dispos. 2018, 46, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.-W.; Xu, X.-H.; Feng, S.-L.; Tang, Z.-B.; Chen, S.-W.; Hui, L. Synthesis of hybrid 4-deoxypodophyllotoxin-5-fluorouracil compounds that inhibit cellular migration and induce cell cycle arrest. Bioorg. Med. Chem. Lett. 2016, 26, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; Guan, X.-W.; Hui, L.; Jin, Y.-X.; Chen, S.-W. Investigation of the anti-angiogenesis effects induced by deoxypodophyllotoxin-5-FU conjugate C069 against HUVE cells. Bioorg. Med. Chem. Lett. 2017, 27, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Fu, J.; Tang, Y.; Gao, Y.; Zhang, S.; Guo, Q. Design and synthesis of novel 4’-demethyl-4-deoxypodophyllotoxin derivatives as potential anticancer agents. Bioorg. Med. Chem. Lett. 2016, 26, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Khaled, M.; Jiang, Z.-Z.; Zhang, L.-Y. Deoxypodophyllotoxin: A promising therapeutic agent from herbal medicine. J. Ethnopharmacol. 2013, 149, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, Y.; Zhong, Z.-Y.; Zhang, J.; Li, F.; Jia, L.-L.; Liu, L.; Zhu, X.; Liu, X.-D. Validated LC–MS/MS assay for quantitative determination of deoxypodophyllotoxin in rat plasma and its application in pharmacokinetic study. J. Pharm. Biomed. Anal. 2014, 88, 410–415. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Biological Activity (Tested Model) | Level of Activity a (Control/Mechanism b) | Ref. |
|---|---|---|---|
| 1a | Antitumor (PC-3, SK-OV-3, MDA-MB-231 and MCF-7 cell lines) | IC50 = 0.7–1.2 μM (IC50 = 5.2–24.5 μM to 5-FU) | [33] |
| 1b | Antibacterial (Staphylococcus aureus Newman) | MIC = 0.63–1.2 μM (MIC = 0.54–1.1 μM to vancomycin) | [34] |
| 2 | Antitumor (HepG2 cell line) | IC50 = 39.8 μM (low cytotoxicity to L-02 cell line) | [42] |
| Neuroprotective (hippocampal neurons from mice) | At 10 μM cause ↑ calcium intracellular) | [45] | |
| 2a | Antiviral (Dengue Virus type 2) | EC50 = 1.4 µM with SI = 57.7 (EC50 = 13.5 µM to ribavirin) | [47] |
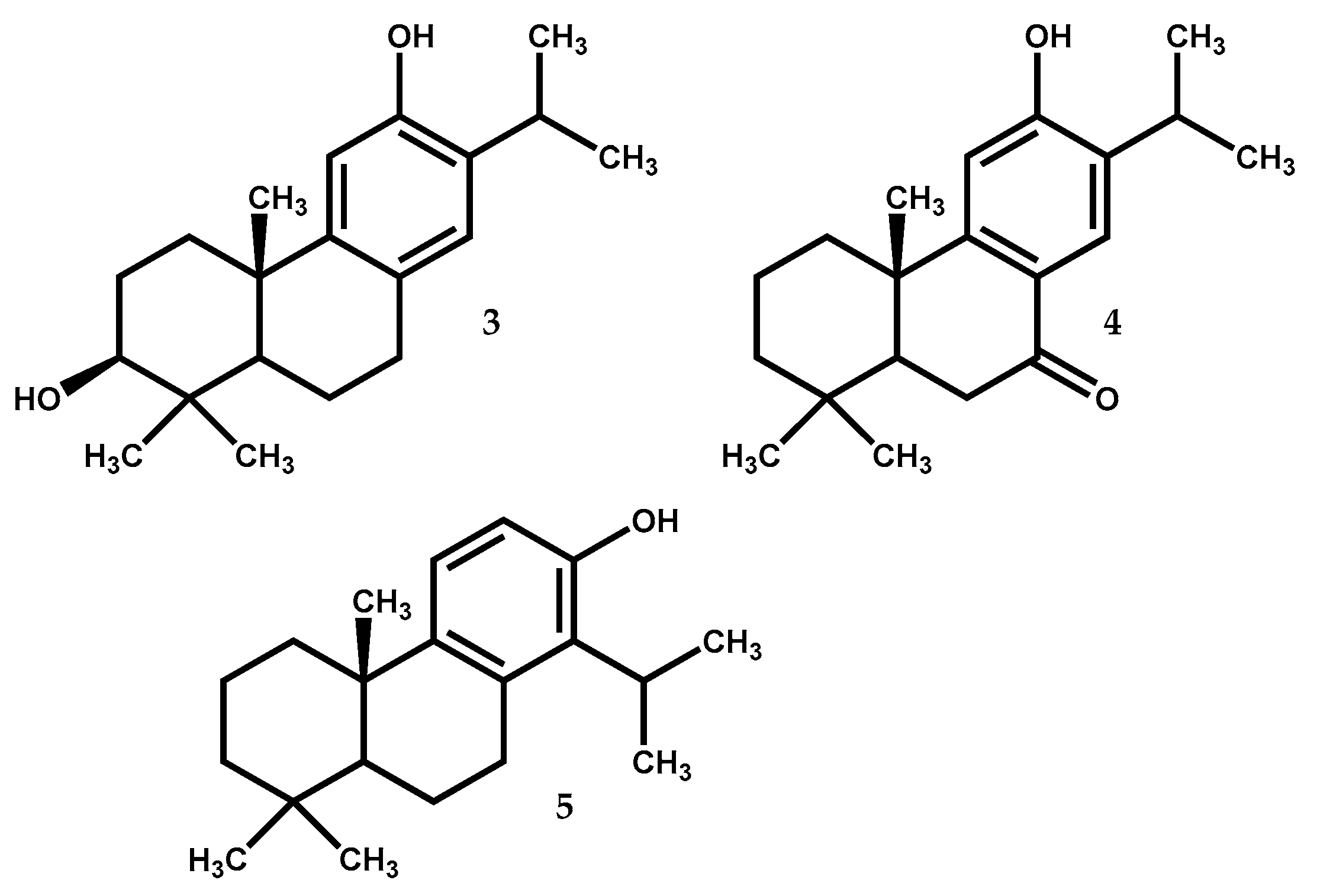
| 3 | Neurons excitability (rat hippocampal CA1 neurons) | At 30 µM cause inhibition of VGSC | [53] |
| 4 | Antidiabetic (α–glucosidase inhibition) | At 33.2 mM cause more than 65% of inhibition (at 15.4 mM acarbose cause identical inhibition) | [57] |
| Antimelanogenesis (tyrosinase inhibition) | At 1.7 mM cause more than 65% of inhibition (at 3.5 mM kojic acid cause identical inhibition) | [57] | |
| Antiviral (MDCK cell line exposed to H1N1 virus) | At 1.7 mM protect against severe cytopathic effect caused by H1N1 virus | [58] | |
| Antitumor (DU145 cell line) | At 20 μM the STAT3 activation was 40% inhibited | [59] | |
| Antitumor (Mia-PaCa2 cell line) | IC50 = 15 μM (↑ Bax expression, ↑ ROS–mediated alterations, ↓ Bcl–2 expression, ↓ migratory capacity | [62] | |
| 5 | Antibacterial (Bacillus subtilis) | At IC50 = 1.5 μM inhibition of metabolic dehydrogenases | [72] |
| Vascular-protection (rats) | At 1–10 μg/kg ↓ infarct volume, ↑ ischemia–induced neurological deficit by activation of PKB/HO–1, SOD and GSH | [73] | |
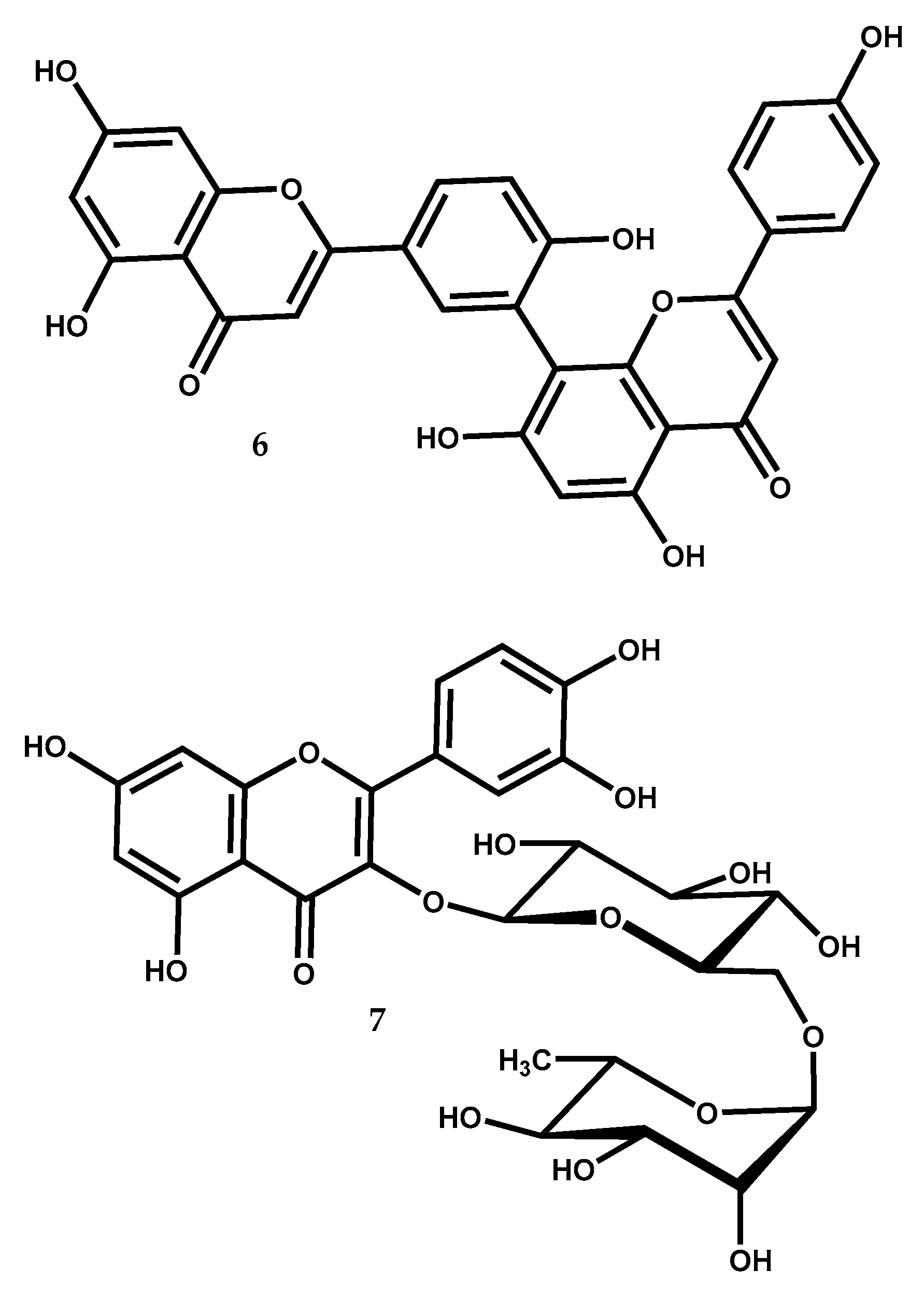
| 6 | Antitumor (aromatase inhibition) | IC50 = 93.6 μM | [91] |
| Antitumor (OVCAR-3 and SK-OV-3) | 20–50 μM cause ↓ cell propagation, block cell cycle progression at the G1/G0 phase and induce cell apoptosis | [94] | |
| Antitumor (A549) | IC50 = 1.54 μM (inhibition of human AKR1B10 activity) | [95] | |
| Anti-arthritis (adjuvant induced arthritic rats) | At 20–40 mg/kg cause ↓ inflammation | [92] | |
| Antidiabetic (DPP-IV inhibition) | IC50 = 3.9 μM | [99] | |
| 7 | Cardioprotective (rat cardiomyoblasts H9c2) | At 50 μM exhibits an apoptosis rate of 20% after pirarubicin–induced toxicity (30% to dexrazoxane) | [103] |
| Antidepressant (in mice) | At 100 mg/kg alleviate CUS | [104] | |
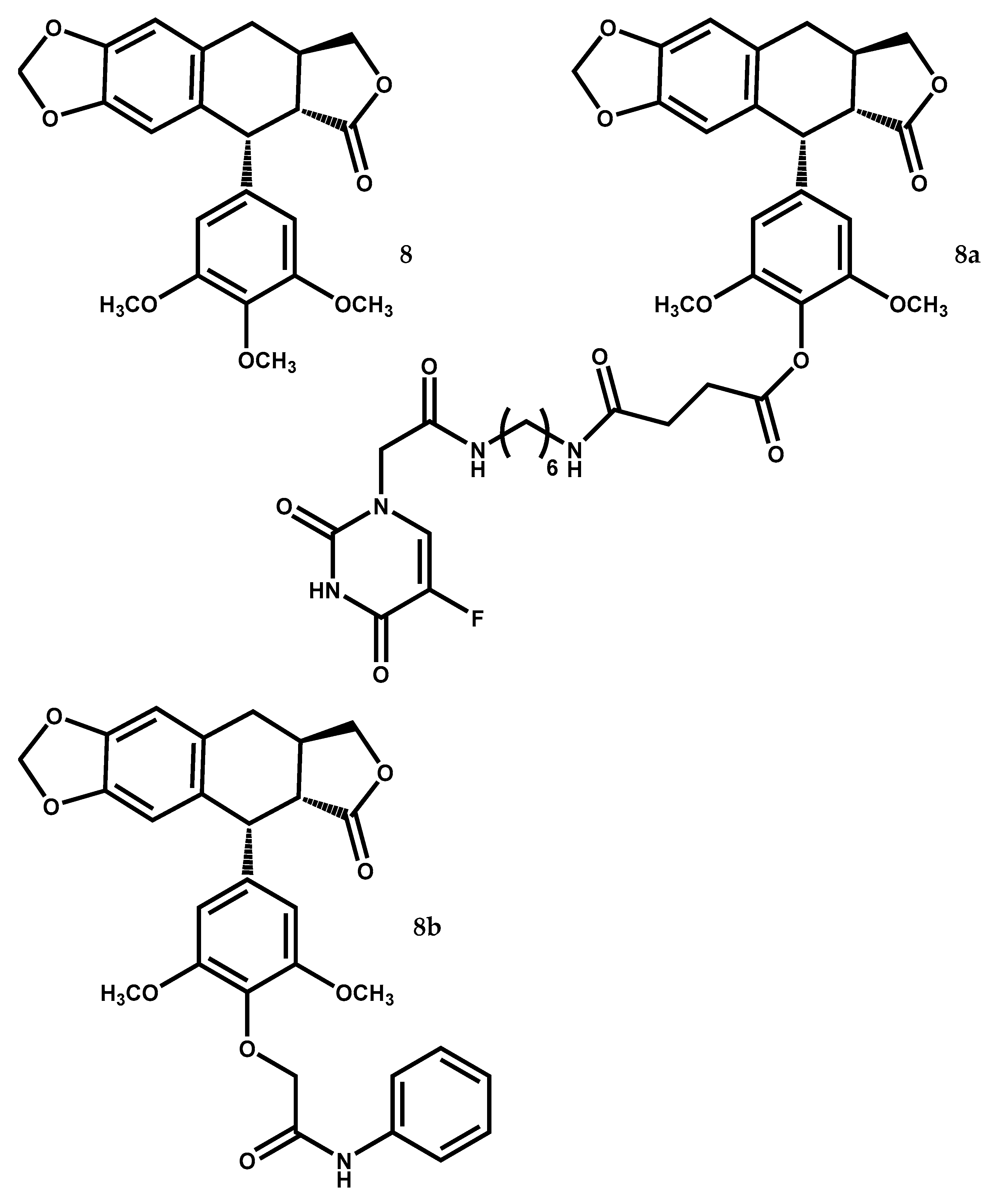
| 8 | Antitumor (U-87 MG, SF126, SGC-7901, BGC-823, HO-8910, SK-0V-3, HT-29, MDA-MB-231, JeG-3) | IC50 = 13.95–26.72 nM by ↓ Cdc2 expression, ↓ cyclin B1, ↓ Cdc25C (IC50 ≥ 73.57 nM to etoposide) | [114] |
| Antitumor (MCF-7/S, MCF-7/A) | IC50 = 5.86 nM, RI = 0.552 (paclitaxel and etoposide exhibit higher IC50 and RI) | [115] | |
| Antitumor (MCF-7/S and in MCF-7/A xenograft mice) | At 12.5 mg/kg 49.2% of tumour volume growth inhibition (identical to paclitaxel) | [115] | |
| 8a | Antitumor (HeLa, A549, HCT-8 and HepG2 cell lines) | IC50 = 0.27–4.03 μM, cell migration inhibition, ↑ TIMP-1 expression, ↓ MMP-9 expression, more selectivity than 5-Fu and etoposide | [128] |
| Antitumor (HUVEC cell line) | At 0.1–0.3 μM higher activity and selectivity index than etoposide at 1 μM | [129] | |
| 8b | Antitumor (MGC-803 cell line) | IC50 = 0.22 μM (IC50 values > 10 μM to etoposide) | [130] |
| Antitumor (HepG2 xenografts in mice) | At 4 mg/kg ↓ in 45.56% the weights and volumes of tumor | [130] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavares, W.R.; Seca, A.M.L. The Current Status of the Pharmaceutical Potential of Juniperus L. Metabolites. Medicines 2018, 5, 81. https://doi.org/10.3390/medicines5030081
Tavares WR, Seca AML. The Current Status of the Pharmaceutical Potential of Juniperus L. Metabolites. Medicines. 2018; 5(3):81. https://doi.org/10.3390/medicines5030081
Chicago/Turabian StyleTavares, Wilson R., and Ana M. L. Seca. 2018. "The Current Status of the Pharmaceutical Potential of Juniperus L. Metabolites" Medicines 5, no. 3: 81. https://doi.org/10.3390/medicines5030081
APA StyleTavares, W. R., & Seca, A. M. L. (2018). The Current Status of the Pharmaceutical Potential of Juniperus L. Metabolites. Medicines, 5(3), 81. https://doi.org/10.3390/medicines5030081