A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders



Abstract
:1. Introduction
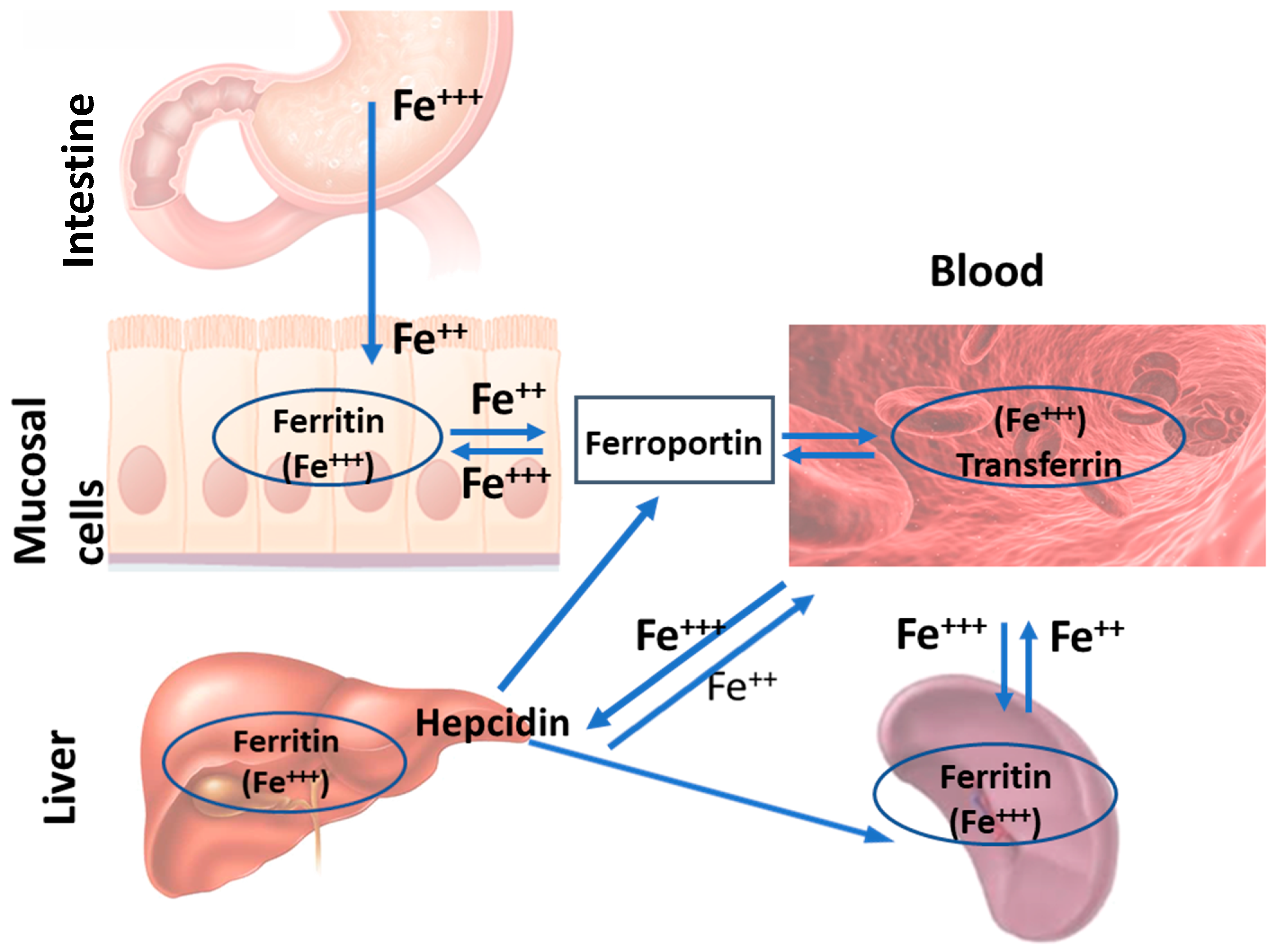
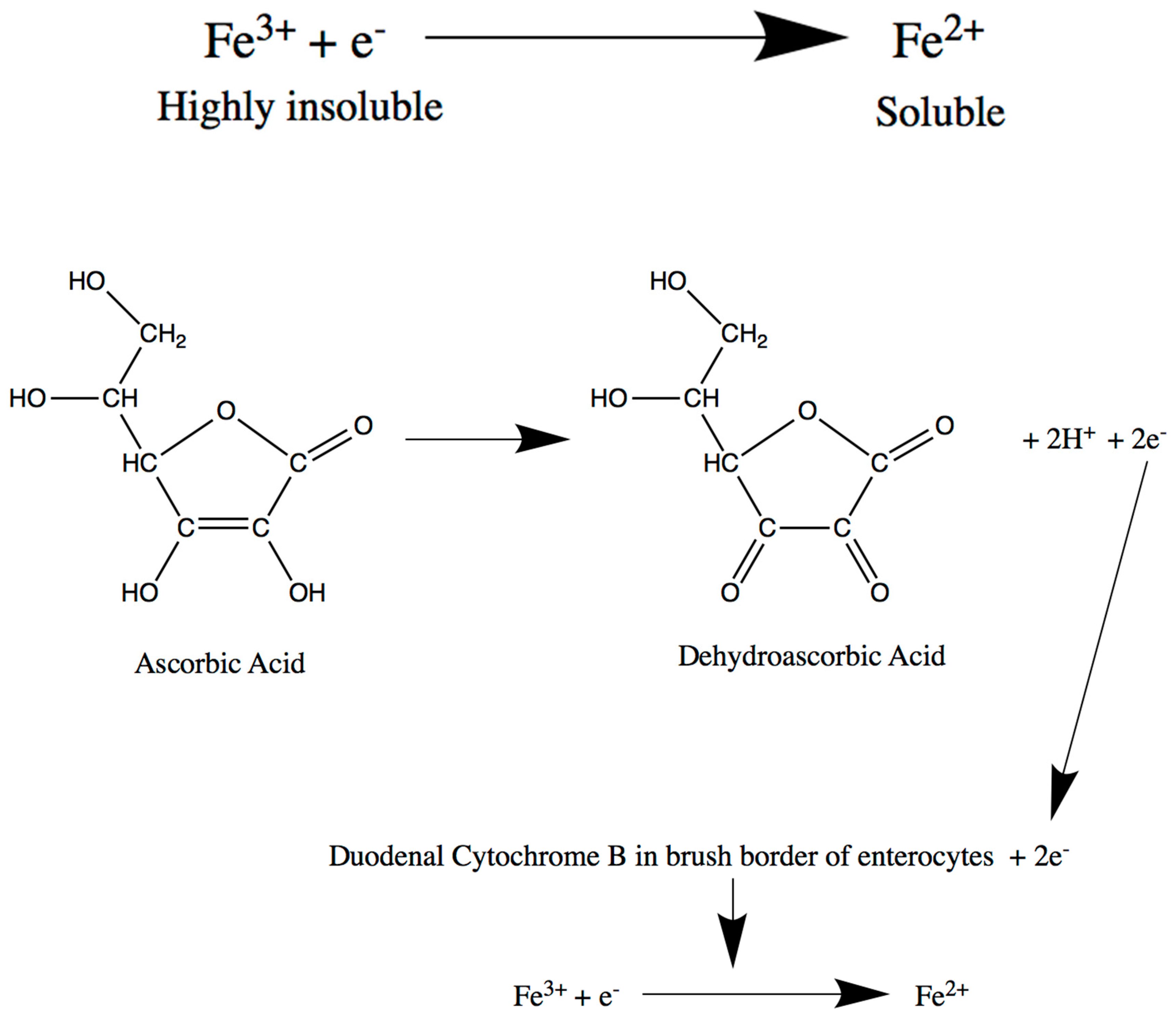
2. Intestine
Intestine-Related Iron Metabolism Disorders
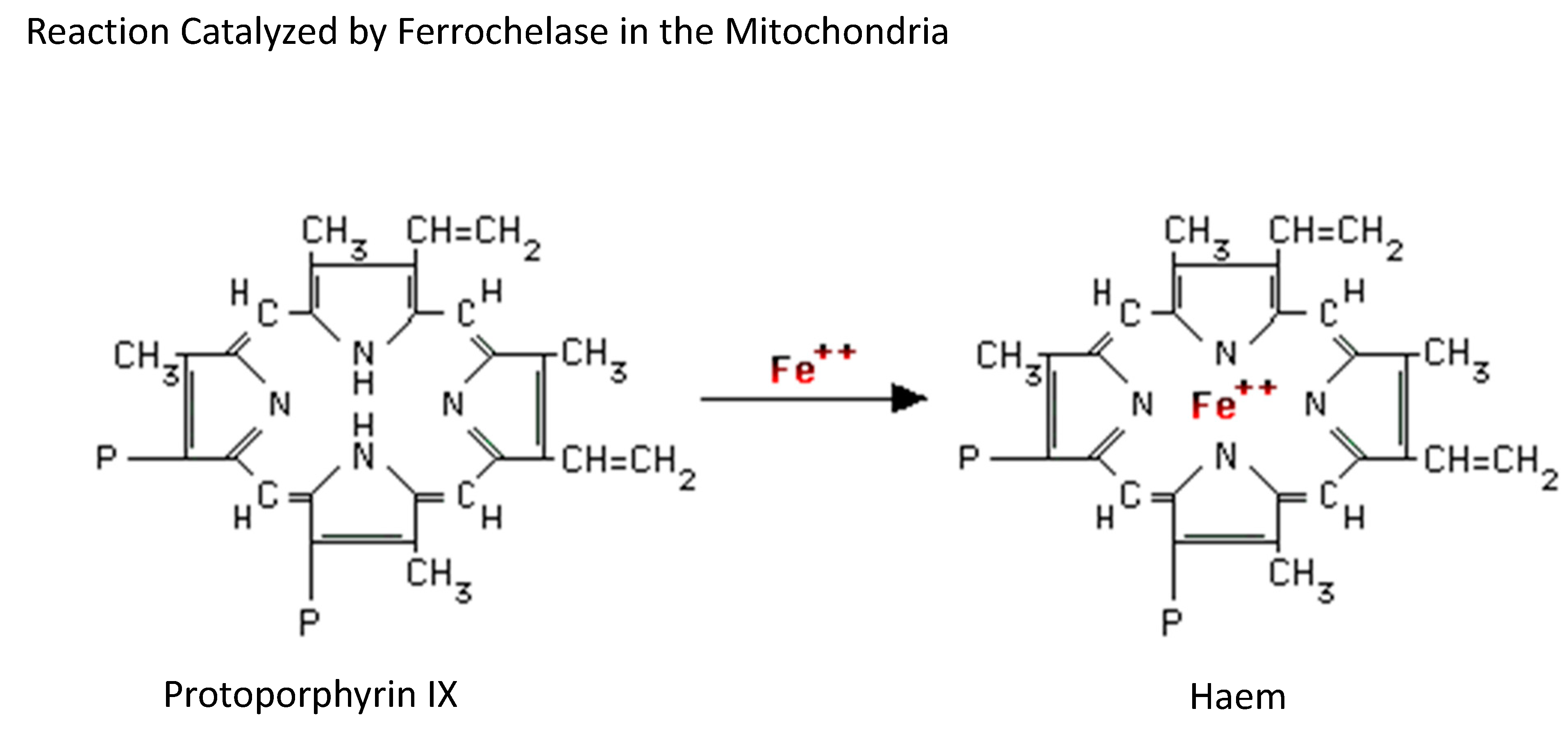
3. Bone Marrow
Bone Marrow-Related Iron Metabolism Disorders
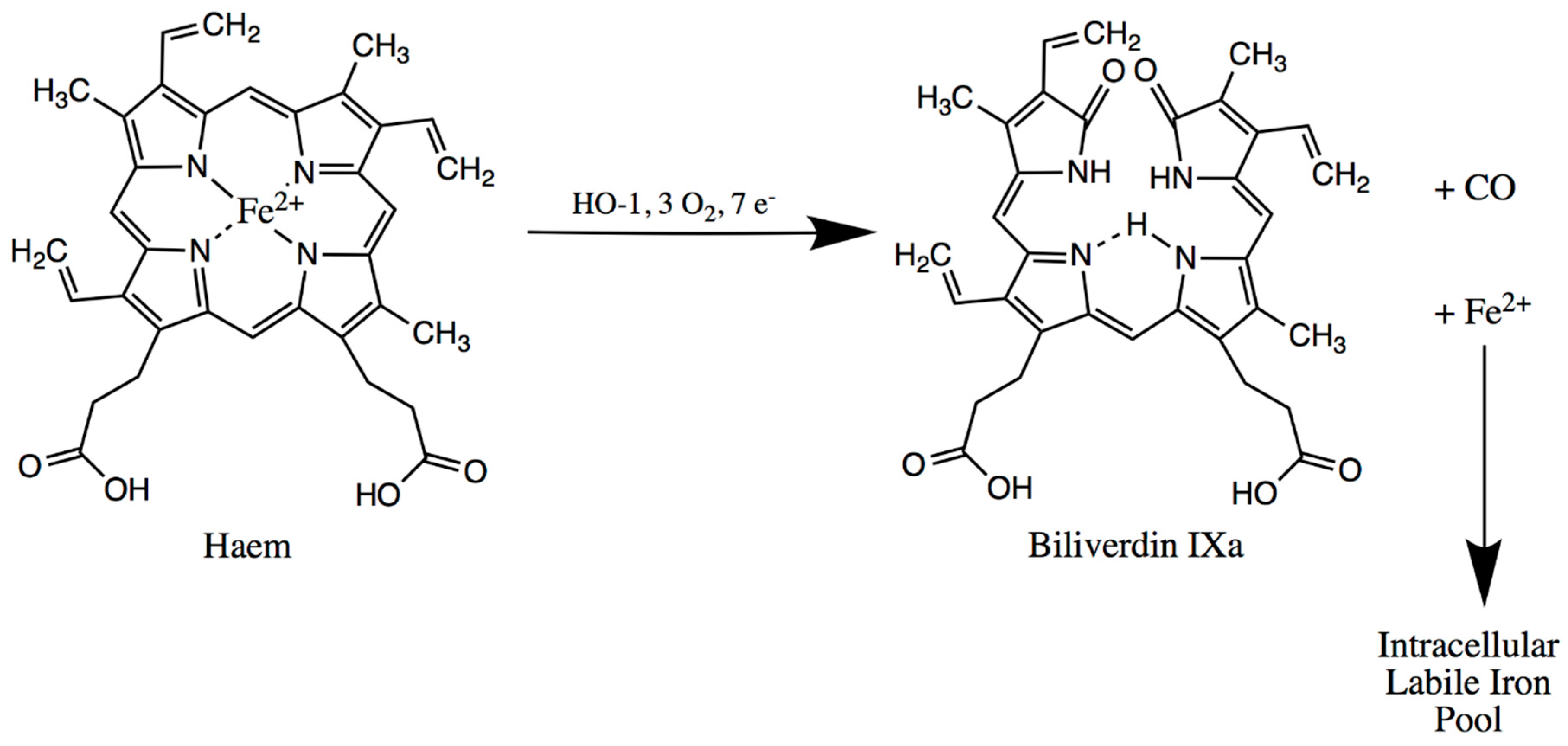
4. Spleen
Spleen-Related Iron Metabolism Disorders
5. Liver
Liver-Related Iron Metabolism Disorders
6. Management
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pantopoulos, K.; Porwal, S.K.; Tartakoe, A.; Devireddy, L. Mechanisms of Mammalian Iron Homeostasis. Biochemistry 2012, 51, 705–5724. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Garcia-Erce, A.J.; Remacha, F.A. Disorders of iron metabolism. Part 1: Molecular basis of iron homoeostasis. J. Clin. Pathol. 2011, 64, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldvogel-Abramowski, S.; Waeber, G.; Gassner, C.; Buser, A.; Frey, B.M.; Favrat, B.; Tissot, J.D. Physiology of Iron Metabolism. Transfus. Med. Hemother. 2014, 41, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Hooda, J.; Shah, A.; Zhang, L. Heme, an Essential Nutrient from Dietary Proteins, Critically Impacts Diverse Physiological and Pathological Processes. Nutrients 2014, 6, 1080–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayeghi, M.; Latunde-Dada, G.; Oakhill, J.; Laftah, A.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Clark, M. Kumar & Clark’s Clinical Medicine, 9th ed.; Elsevier: Edinburgh, UK, 2016. [Google Scholar]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastroint. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [Green Version]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Luo, X.; Hill, M.; Johnson, A. Latunde-Dada GO. Modulation of Dcytb (Cybrd 1) expression and function by iron, dehydroascorbate and Hif-2 alpha in cultured cells. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 106–112. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.; Gunshin, Y.; Romero, M.; Boron, W.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Chen, H.; Attieh, Z.; Su, T.; Syed, B.; Gao, H.; Alaeddine, R.; Fox, T.C.; Usta, J.; Naylor, C.E.; Evans, R.W.; et al. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood 2004, 103, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar] [PubMed]
- Miller, J.L. Iron Deficiency Anemia: A Common and Curable Disease. Cold Spring Harb. Perspect. Med. 2013, 3, a011866. [Google Scholar] [CrossRef] [PubMed]
- Marignani, M.; Angeletti, S.; Bordi, C.; Malagnino, F.; Mancino, C.; DelleFave, G.; Annibale, B. Reversal of long-standing iron deficiency anaemia after eradication of Helicobacter pylori infection. Scand. J. Gastroenterol. 1997, 32, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Garcia-Erce, A.J.; Remacha, F.A. Disorders of iron metabolism. Part II: Iron deficiency and iron overload. J. Clin. Pathol. 2011, 64, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P. Transferrin receptor 1. Int. J. Biochem. Cell Biol. 2004, 36, 2137–2143. [Google Scholar] [CrossRef]
- Bali, P.; Zak, O.; Aisen, P. A New Role for the Transferrin Receptor in the Release of Iron from Transferrin. Biochemistry 1991, 30, 324–328. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Regulation of iron acquisition and storage: Consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81. [Google Scholar] [CrossRef]
- Dautry-Varsat, A. Receptor-mediated endocytosis: The intracellular journey of transferrin and its receptor. Biochimie 1986, 68, 375–381. [Google Scholar] [CrossRef]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H. pH and the Recycling of Transferrin during Receptor-Mediated Endocytosis. Proc. Natl. Acad. Sci. USA-Biol. Sci. 1983, 80, 2258–2262. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Nai, A.; Lidonnici, M.R.; Rausa, M.; Mandelli, G.; Pagani, A.; Silvestri, L.; Ferrari, G.; Camaschella, C. The second transferrin receptor regulates red blood cell production in mice. Blood 2015, 125, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Khalil, S.; Holy, M.; Grado, S.; Fleming, R.; Kurita, R.; Nakamura, Y.; Goldfarb, A. A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2. Blood Adv. 2017, 1, 1181–1194. [Google Scholar] [CrossRef] [Green Version]
- Troadec, M.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted deletion of the mouse Mitoferrin1 gene: From anemia to protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef]
- Knutson, M.; Wessling-Resnick, M. Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef]
- Kuypers, F.A.; De Jong, K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell. Mol. Biol. 2004, 50, 147–158. [Google Scholar]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [Green Version]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B., Jr.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, M.; Graversen, J.; Jacobsen, C.; Sonne, O.; Hoffman, H.; Law, S.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.; Jacobsen, C.; Hojrup, P.; Moller, H.; Moestrup, S. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Park, C.; Valore, E.; Waring, A.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loréal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef]
- Kulaksiz, H.; Fein, E.; Redecker, P.; Stremmel, W.; Adler, G.; Cetin, Y. Pancreatic beta-cells express hepcidin, an iron-uptake regulatory peptide. J. Endocrinol. 2008, 197, 241–249. [Google Scholar] [CrossRef]
- Kulaksiz, H.; Theilig, F.; Bachmann, S.; Gehrke, S.; Rost, D.; Janetzko, A.; Cetin, Y.; Stremmel, W. The iron-regulatory peptide hormone hepcidin: expression and cellular localization in the mammalian kidney. J. Endocrinol. 2005, 184, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Valore, E.V.; Ganz, T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol. Dis. 2008, 40, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.; Powelson, J.; Vaughn, M.; Donovan, A.; Ward, D.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; del-Castillo-Rueda, A.; Moran-Jimenez, M.; Ganz, T.; Nemeth, E. Hepcidin-Induced Endocytosis of Ferroportin Is Dependent on Ferroportin Ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Gao, J.; Koeberl, D.D.; Enns, C.A. The Role of Hepatocyte Hemojuvelin in the Regulation of Bone Morphogenic Protein-6 and Hepcidin Expression in Vivo. J. Biol. Chem. 2010, 285, 16416–16423. [Google Scholar] [CrossRef] [Green Version]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 2009, 87, 471–480. [Google Scholar] [CrossRef]
- Enns, C.A.; Ahmed, R.; Wang, J.; Ueno, A.; Worthen, C.; Tsukamoto, H.; Zhang, A.S. Increased Iron Loading Induces Bmp6 Expression in the Non-Parenchymal Cells of the Liver Independent of the BMP-Signaling Pathway. PLoS ONE 2013, 8, e60534. [Google Scholar] [CrossRef]
- Gross, C.; Irrinki, A.; Feder, J.; Enns, C. Co-trafficking of HFE, a nonclassical major histocompatibility complex class I protein, with the transferrin receptor implies a role in intracellular iron regulation. J. Biol. Chem. 1998, 273, 22068–22074. [Google Scholar] [CrossRef]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.; Enns, C.A. Interaction of the Hereditary Hemochromatosis Protein HFE with Transferrin Receptor 2 Is Required for Transferrin-Induced Hepcidin Expression. Cell Metab. 2009, 9, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.J.; Fleming, M.D. Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am. J. Hematol. 2012, 87, 588–595. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Wu, Q.; Cheng, W.; Liu, W.; Zhao, Y.; Mayeur, C.; Schmidt, P.J.; Yu, P.B.; Wang, F.; et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 2014, 124, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Wrighting, D.M.; Andrews, N.C. Interleukin induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef] [Green Version]
- Steinbicker, A.U.; Muckenthaler, M.U. Out of balance—Systemic iron homeostasis in iron-related disorders. Nutrients 2013, 5, 3034–3061. [Google Scholar] [CrossRef]
- Imlay, J.; Chin, S.; Linn, S. Toxic DNA Damage by Hydrogen-Peroxide through the Fenton Reaction in Vivo and in Vitro. Science 1988, 240, 640–642. [Google Scholar] [CrossRef]
- Pietrangelo, A. Mechanisms of iron hepatotoxicity. J. Hepatol. 2016, 65, 226–227. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
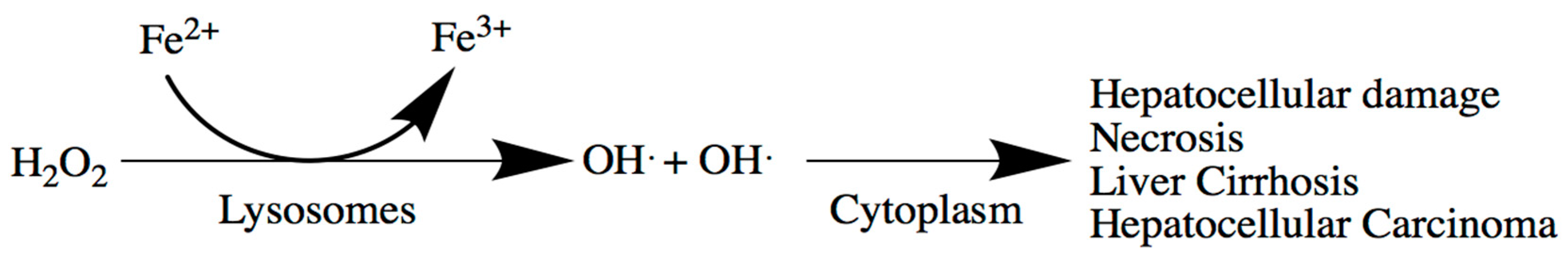{kind=link}
| Disorder | Features and Symptoms | Management |
|---|---|---|
| Peptic ulcers | Abdominal pain, nausea, vomiting, loss of appetite, weight loss, gastric/duodenal bleeding, anaemia. | Treatment of the underlying condition; iron supplementation; blood transfusion in severe cases. |
| Inflammatory Bowel Disease | Diarrhoea, low-grade fever, abdominal pain, blood in the stool, reduced appetite, weight loss, anaemia. | Corticosteroids, immunosuppressants. |
| Hookworm infection | Abdominal pain, fever, nausea, blood in the stool, anaemia. | Antihelminthic drugs (mebendazole); iron supplementation if anaemic. |
| Malabsorption syndromes | Diarrhoea, steatorrhoea, weight loss and fatigue, oedema, anaemia. | Gluten-free diet; iron and vitamin supplementation. |
| Thalassaemia syndromes | Skin pallor, jaundice, slowed growth, splenomegaly, facial bone deformities; transfusion-dependent iron overload. | Blood transfusions; iron chelation (deferoxamine, deferasirox) for secondary haemochromatosis; folic acid supplementation. |
| Sickle cell anaemia | Sickle cell crisis (abdominal pain, bone pain); anaemia; pallor, jaundice, hepatomegaly, splenomegaly in children followed by splenic infarction later, chronic leg ulcers; prone to pneumococcal septicaemia. | Prophylactic management (avoid precipitating factors); folic acid supplementation; in case of crisis: rehydration, antibiotics, and bicarbonate if acidotic; blood transfusion only if severe anaemia. |
| Sideroblastic anaemia | Skin pallor, fatigue, dizziness, splenomegaly; transfusion-dependent iron overload. | High-dose pyridoxine, folic acid supplementation, blood transfusion if severe anaemia; iron chelation (deferoxamine) if transfusion-related iron overload. |
| Anaemia of chronic disease | Tiredness, weakness, dyspnoea, skin pallor, dizziness. | Treatment of underlying cause if possible; iron supplementation is not beneficial; blood transfusion in severe anaemia. |
| Haemochromatosis | Joint pain, abdominal pain, fatigue, weakness, impotence; cirrhosis, diabetes mellitus, skin hyperpigmentation, heart failure. | Therapeutic phlebotomy; iron chelation therapy. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yiannikourides, A.; Latunde-Dada, G.O. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. https://doi.org/10.3390/medicines6030085
Yiannikourides A, Latunde-Dada GO. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines. 2019; 6(3):85. https://doi.org/10.3390/medicines6030085
Chicago/Turabian StyleYiannikourides, Andronicos, and Gladys O. Latunde-Dada. 2019. "A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders" Medicines 6, no. 3: 85. https://doi.org/10.3390/medicines6030085
APA StyleYiannikourides, A., & Latunde-Dada, G. O. (2019). A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines, 6(3), 85. https://doi.org/10.3390/medicines6030085