
Stem Cell Therapies for Epidermolysis Bullosa Treatment



Abstract
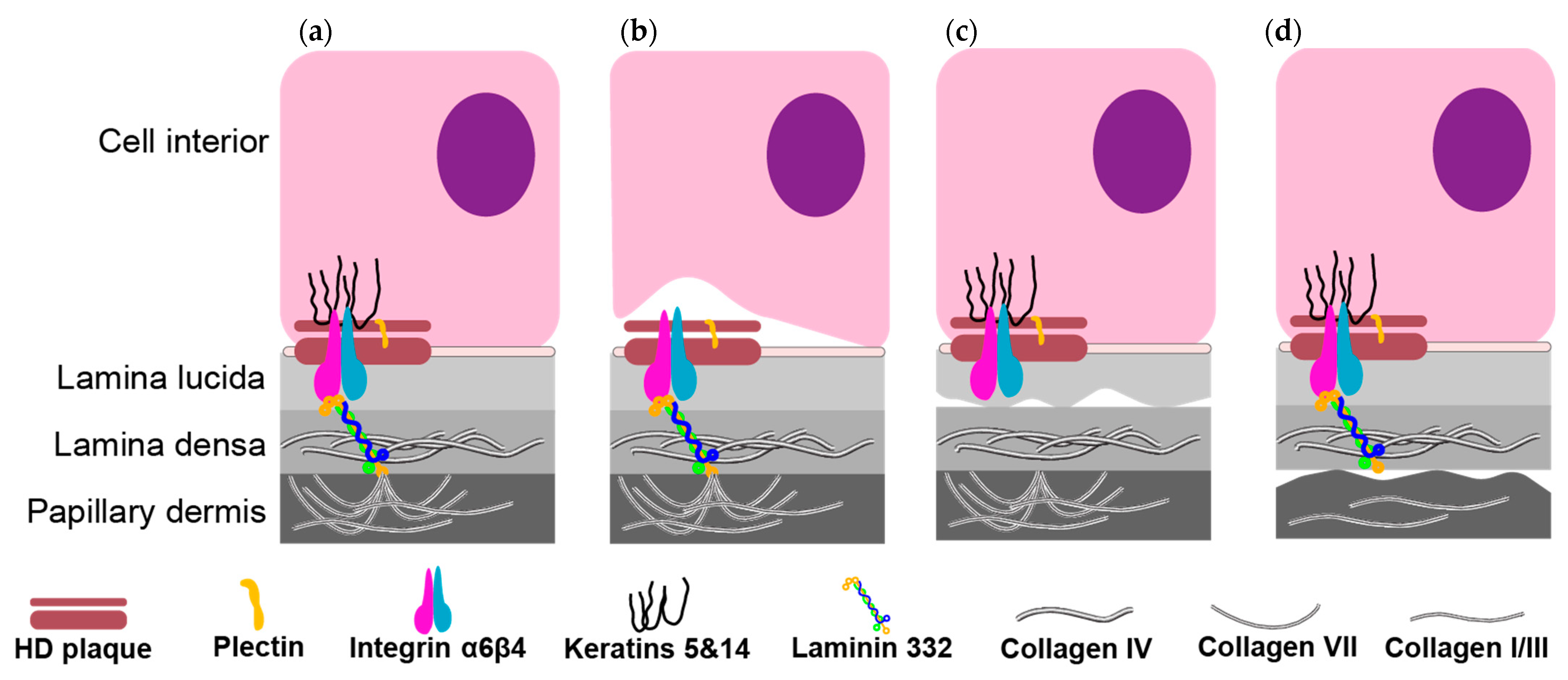
:1. Introduction
2. Types of Epidermolysis Bullosa
2.1. Epidermolysis Bullosa Simplex (EBS)
2.2. Junctional Epidermolysis Bullosa (JEB)
2.3. Dystrophic Epidermolysis Bullosa (DEB)
2.4. Mixed Epidermolysis Bullosa—Kindler Syndrome
3. Introduction to Stem Cells
- Totipotent Cells: They have the ability to differentiate into all possible cell types. For example, a fertilized oocyte and the first few cells that result from the division of the zygote are totipotent cells [32].
- Pluripotent Cells: They have the ability to differentiate into cells that come from the 3 germ layers—ectoderm, endoderm, and mesoderm—from which all tissues and organs derive [33]. Examples include embryonic stem cells (ESCs), as well as induced pluripotent stem cells (iPSCs) which arise from reprogramming somatic cells [34].
- Oligopotent Cells: They have the ability to differentiate into a few lineages within a single tissue. This category includes hematopoietic stem cells (HSCs) which can differentiate into lymphoid or myeloid stem cells [38].
- Unipotent Cells: They have the ability to only produce cells of their own type, such as muscle stem cells which can differentiate into only mature muscle cells [39].
- As for the classification based on their origin, stem cells can be classified as below:
- Adult Stem Cells: they come from the “niches” in adult tissues. They are characterized as multipotent because they can self-renew, but have a low ability to differentiate. They can mature in cells of the tissue or organ from which they originate but also play a key role in their maintenance and repair [40].
- Advanced Perinatal embryonic stem cells: Perinatal stem cells (e.g., from aborted embryos or umbilical cord blood). They are the various types of stem cells found in the umbilical cord unit, such as hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), amniotic epithelial cells (AMSCs), chorionic mesenchymal stem cells (CMSCs) and progenitor endothelial stem cells, as well as earlier cell types such as unrestricted somatic stem cells (USSCs) and very small embryonic-like cells (Very Small Embryonic Like Stem Cells, VSELs). The majority of the scientific community characterizes them as pluripotent and not simply as multipotent cells, which gives them the characteristic of the so-called plasticity of these stem cells and has led to a wide range of clinical applications [41].
- Embryonic stem cells: Embryonic cells that arise during the first four days after fertilization are considered totipotent and form all the cells of the embryo as well as extraembryonic formations, such as the placenta. Embryonic stem cells that arise after four to five days are considered pluripotent, they come from inside the blastocyst and can transform into all cell types of an organism [42]. A third class of “embryonic” cells, so-called induced pluripotent cells (iPSCs), has been added in recent years. iPSCs are developed through the genetic manipulation of differentiated cells [43].
4. Cellular Therapy Options for EB
4.1. Hematopoietic Stem Cells (HSCs) Transplantation
4.2. Cytotherapies with Mesenchymal Stem Cells (MSCs)
4.3. Genetically Corrected Autologous Epidermal Stem Cell and Fibroblasts Therapies
4.4. Induced Pluripotent Stem Cells (iPSs)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EB | Epidermolysis bullosa |
| EBS | Epidermolysis bullosa simplex |
| ECM | extracellular matrix |
| JEB | Epidermolysis bullosa conjunctiva |
| JEB-H | Epidermolysis Bullosa Herlitz |
| JEB-nH | Epidermolysis bullosa non-Herlitz |
| LOC syndrome | Laryngo-onycho-cutaneous syndrome |
| DEB | Dystrophic epidermolysis bullosa |
| DDEB | Autosomal dominant dystrophic epidermolysis bullosa |
| RDEB | Autosomal recessive dystrophic epidermolysis bullosa |
| MSCs | Mesenchymal stem cells |
| HCs | hematopoietic stem cells |
| BM | Bone marrow |
| BMT | Bone marrow transplantation |
| iPSs | Induced pluripotent stem cells |
| COLVII | Collagen type VII |
References
- Kanitakis, J. Anatomy, histology and immunohistochemistry of normal human skin. Eur. J. Dermatol. 2002, 12, 390–401. [Google Scholar]
- Gawkrodger, D.J. Dermatology—An Illustrated Colour Text, 3rd ed.; Churchill Livingstone: London, UK, 2002. [Google Scholar]
- Breitkreutz, D.; Koxholt, I.; Thiemann, K.; Nischt, R. Skin Basement Membrane: The Foundation of Epidermal Integrity—BM Functions and Diverse Roles of Bridging Molecules Nidogen and Perlecan. Biomed. Res. Int. 2013, 2013, 179784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurchenco, P.D. Basement Membranes: Cell Scaffoldings and Signaling Platforms. Cold Spring Harb. Perspect Biol. 2011, 3, a004911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aumailley, M.; Rousselle, P. Laminins of the dermo-epidermal junction. Matrix. Biol. 1999, 18, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Borradori, L.; Sonnenberg, A. Structure and function of hemidesmosomes: More than simple adhesion complexes. J. Investig. Dermatol. 1999, 112, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Sawamura, D.; Li, K.H.; Nomura, K.; Sugita, Y.; Christiano, A.M.; Uitto, J. Bullous pemphigoid antigen: cDNA cloning, cellular expression, and evidence for polymorphism of the human gene. J. Invest. Dermatol. 1991, 96, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Carter, W.G.; Ryan, M.C.; Gahr, P.J. Epiligrin, a new cell adhesion ligand for integrin α3β1 in epithelial basement membranes. Cell 1991, 65, 599–610. [Google Scholar] [CrossRef]
- Rousselle, P.; Lunstrum, G.P.; Keene, D.R.; Burgeson, R.E. Kalinin: An epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J. Cell Biol. 1991, 114, 567–576. [Google Scholar] [CrossRef]
- Murat-Sušić, S.; Husar, K.; Skerlev, M.; Marinović, B.; Babić, I. Inherited epidermolysis bullosa—The spectrum of complications. Acta Derm. Croat. 2011, 19, 255–263. [Google Scholar]
- Salam, A.; Proudfoot, L.E.; McGrath, J.A. Inherited blistering skin diseases: Underlying molecular mechanisms and emerging therapies. Ann. Med. 2014, 46, 49–61. [Google Scholar] [CrossRef]
- Köbner, H. Hereditäre Anlage zur Blasenbildung (Epidermolysis bullosa hereditaria). Aus. Poliklin. 1886, 12, 21–22. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.W. Studies on the pathogenesis of Epidermolysis bullosa. J. Invest. Dermatol. 1962, 39, 551–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, J.A. Recently Identified Forms of Epidermolysis Bullosa. Ann. Dermatol. 2015, 27, 658–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, G.B.; Meara, R.H. Epidermolysis bullosa resembling juvenile dermatitis herpetiformis. Br. J. Dermatol. 1954, 66, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Kerns, M.L.; Fuchs, E. Epidermolysis bullosa simplex: A paradigm for disorders of tissue fragility. J. Clin. Invest. 2009, 119, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Rousselle, P.; Beck, K. Laminin 332 processing impacts cellular behavior. Cell. Adh. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Rousselle, P.; Michopoulou, A. Laminin 332 in Junctional Epidermolysis and as an Autoantigen in Mucous Membrane Pemphigoid. In Blistering Disease; Springer: Cham, Switzerland, 2015; pp. 91–102. Available online: http://link.springer.com/10.1007/978-3-662-45698-9_9 (accessed on 25 February 2023).
- McLean, W.H.I.; Irvine, A.D.; Hamill, K.J.; Whittock, N.V.; Coleman-Campbell, C.M.; Mellerio, J.E.; Ashton, G.S.; Dopping-Hepenstal, P.J.H.; Eady, R.A.J.; Jamil, T.; et al. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum. Mol. Genet. 2003, 12, 2395–2409. [Google Scholar] [CrossRef]
- Bruckner-Tuderman, L.; Höpfner, B.; Hammami-Hauasli, N. Biology of anchoring fibrils: Lessons from dystrophic epidermolysis bullosa. Matrix. Biol. 1999, 18, 43–54. [Google Scholar] [CrossRef]
- Dang, N.; Murrell, D.F. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp. Dermatol. 2008, 17, 553–568. [Google Scholar] [CrossRef]
- Nyström, A.; Bruckner-Tuderman, L.; Kiritsi, D. Dystrophic Epidermolysis Bullosa: Secondary Disease Mechanisms and Disease Modifiers. Front. Genet. 2021, 12, 1737. [Google Scholar] [CrossRef] [PubMed]
- Mariath, L.M.; Santin, J.T.; Schuler-Faccini, L.; Kiszewski, A.E. Inherited epidermolysis bullosa: Update on the clinical and genetic aspects. An. Bras. Dermatol. 2020, 95, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfendner, E.G.; Lucky, A.W. Dystrophic Epidermolysis Bullosa; Director, EBDx Program, GeneDx, Inc.: Gaithersburg, MD, USA; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Bruckner-Tuderman, L. Dystrophic epidermolysis bullosa: Pathogenesis and clinical features. Derm. Clin. 2010, 28, 107–114. [Google Scholar] [CrossRef]
- Intong, L.R.A.; Murrell, D.F. Inherited epidermolysis bullosa: New diagnostic criteria and classification. Clin. Dermatol. 2012, 30, 70–77. [Google Scholar] [CrossRef]
- Fine, J.-D.; Johnson, L.B.; Weiner, M.; Li, K.-P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef]
- Has, C.; Castiglia, D.; del Rio, M.; Diez, M.G.; Piccinni, E.; Kiritsi, D.; Kohlhase, J.; Itin, P.; Martin, L.; Fischer, J.; et al. Kindler syndrome: Extension of FERMT1 mutational spectrum and natural history. Hum. Mutat. 2011, 32, 1204–1212. [Google Scholar] [CrossRef] [Green Version]
- Kolios, G.; Moodley, Y. Introduction to Stem Cells and Regenerative Medicine. Respiration 2013, 85, 3–10. [Google Scholar] [CrossRef]
- Rossant, J. Stem cells from the Mammalian blastocyst. Stem Cells 2001, 19, 477–482. [Google Scholar] [CrossRef]
- De Miguel, M.P.; Fuentes-Julián, S.; Alcaina, Y. Pluripotent stem cells: Origin, maintenance and induction. Stem Cell Rev. Rep. 2010, 6, 633–649. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Poniewierska, A.; Suszynska, M.; Ratajczak, J. Pluripotent and multipotent stem cells in adult tissues. Adv. Med. Sci. 2012, 57, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Augello, A.; Kurth, T.B.; De Bari, C. Mesenchymal stem cells: A perspective from in vitro cultures to in vivo migration and niches. Eur. Cell Mater. 2010, 20, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Barzilay, R.; Melamed, E.; Offen, D. Introducing transcription factors to multipotent mesenchymal stem cells: Making transdifferentiation possible. Stem Cells 2009, 27, 2509–2515. [Google Scholar] [CrossRef]
- Marone, M.; De Ritis, D.; Bonanno, G.; Mozzetti, S.; Rutella, S.; Scambia, G.; Pierelli, L. Cell cycle regulation in human hematopoietic stem cells: From isolation to activation. Leuk. Lymphoma 2002, 43, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Rudnicki, M.A. The emerging biology of muscle stem cells: Implications for cell-based therapies. Bioessays 2013, 35, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Quesenberry, P.J.; Dooner, G.; Colvin, G.; Abedi, M. Stem cell biology and the plasticity polemic. Exp. Hematol. 2005, 33, 389–394. [Google Scholar] [CrossRef]
- Abbaspanah, B.; Momeni, M.; Ebrahimi, M.; Mousavi, S.H. Advances in perinatal stem cells research: A precious cell source for clinical applications. Regen. Med. 2018, 13, 595–610. [Google Scholar] [CrossRef]
- Stojkovic, M.; Lako, M.; Strachan, T.; Murdoch, A. Derivation, growth and applications of human embryonic stem cells. Reproduction 2004, 128, 259–267. [Google Scholar] [CrossRef]
- Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 2007, 1, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Nita, M.; Pliszczyński, J.; Eljaszewicz, A.; Moniuszko, M.; Ołdak, T.; Woźniak, K.; Majewski, S.; Kowalewski, C.; Kamiński, A.; Śladowski, D.; et al. Surgical Treatment of Wounds Using Stem Cells in Epidermolysis Bullosa (EB); Valarmathi, M.T., Ed.; IntechOpen: Rijeka, Croatia, 2021. [Google Scholar] [CrossRef]
- Naso, G.; Petrova, A. Cellular therapy options for genetic skin disorders with a focus on recessive dystrophic epidermolysis bullosa. Br. Med. Bull. 2020, 136, 30–45. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, S.-J.; Kim, S.-E.; Kim, K.; Cho, B.; Roh, K.; Kim, S.-C. Intravenous allogeneic umbilical cord blood–derived mesenchymal stem cell therapy in recessive dystrophic epidermolysis bullosa patients. J. Clin. Investig. 2021, 6, 143606. [Google Scholar] [CrossRef]
- Kiritsi, D.; Dieter, K.; Niebergall-Roth, E.; Fluhr, S.; Daniele, C.; Esterlechner, J.; Sadeghi, S.; Ballikaya, S.; Erdinger, L.; Schauer, F.; et al. Clinical trial of ABCB5+ mesenchymal stem cells for recessive dystrophic epidermolysis bullosa. J. Clin. Investig. 2021, 6, 151922. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Yamazaki, T.; Chino, T.; Ishii, M.; Otsuru, S.; Kikuchi, Y.; Iinuma, S.; Saga, K.; Nimura, K.; Shimbo, T.; et al. PDGFRα-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc. Natl. Acad. Sci. USA 2011, 108, 6609–6614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodinger, C.; Reichelt, J.; Bauer, J.W.; Laimer, M. Epidermolysis bullosa: Advances in research and treatment. Exp. Dermatol. 2019, 28, 1176–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.E.; Ishida-Yamamoto, A.; McGrath, J.A.; Hordinsky, M.; Keene, D.R.; Woodley, D.T.; Chen, M.; Riddle, M.J.; Osborn, M.J.; Lund, T.; et al. Bone Marrow Transplantation for Recessive Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 2010, 363, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Ebens, C.; McGrath, J.; Tamai, K.; Hovnanian, A.; Wagner, J.; Riddle, M.; Keene, D.; DeFor, T.; Tryon, R.; Chen, M.; et al. Bone marrow transplant with post-transplant cyclophosphamide for recessive dystrophic epidermolysis bullosa expands the related donor pool and permits tolerance of nonhaematopoietic cellular grafts. Br. J. Dermatol. 2019, 181, 1238–1246. [Google Scholar] [CrossRef]
- Rashidghamat, E.; Kadiyirire, T.; Ayis, S.; Petrof, G.; Liu, L.; Pullabhatla, V.; Ainali, C.; Guy, A.; Aristodemou, S.; McMillan, J.R.; et al. Phase I/II open-label trial of intravenous allogeneic mesenchymal stromal cell therapy in adults with recessive dystrophic epidermolysis bullosa. J. Am. Acad. Dermatol. 2020, 83, 447–454. [Google Scholar] [CrossRef]
- Petrof, G.; Lwin, S.M.; Martinez-Queipo, M.; Abdul-Wahab, A.; Tso, S.; Mellerio, J.E.; Slaper-Cortenbach, I.; Boelens, J.J.; Tolar, J.; Veys, P.; et al. Potential of Systemic Allogeneic Mesenchymal Stromal Cell Therapy for Children with Recessive Dystrophic Epidermolysis Bullosa. J. Invest. Dermatol. 2015, 135, 2319–2321. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, L.; Enzo, E.; Zardi, G.; Bodemer, C.; Magnoni, C.; Schneider, H.; De Luca, M. Hologene 5: A Phase II/III Clinical Trial of Combined Cell and Gene Therapy of Junctional Epidermolysis Bullosa. Front. Genet. 2021, 12, 1507. [Google Scholar] [CrossRef]
- Lwin, S.M.; Syed, F.; Di, W.-L.; Kadiyirire, T.; Liu, L.; Guy, A.; Petrova, A.; Abdul-Wahab, A.; Reid, F.; Phillips, R.; et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. J. Clin. Investig. 2019, 4, e126243. [Google Scholar] [CrossRef] [Green Version]
- Marinkovich, M.; Lane, A.; Sridhar, K.; Keene, D.; Malyala, A.; Maslowski, J. 591 A phase 1/2 study of genetically-corrected, collagen VII expressing autologous human dermal fibroblasts injected into the skin of patients with recessive dystrophic epidermolysis bullosa (RDEB). J. Invest. Dermatol. 2018, 138, S100. [Google Scholar] [CrossRef]
- So, J.Y.; Nazaroff, J.; Iwummadu, C.V.; Harris, N.; Gorell, E.S.; Fulchand, S.; Bailey, I.; McCarthy, D.; Siprashvili, Z.; Marinkovich, M.P.; et al. Long-term safety and efficacy of gene-corrected autologous keratinocyte grafts for recessive dystrophic epidermolysis bullosa. Orphanet J. Rare Dis. 2022, 17, 377. [Google Scholar] [CrossRef]
- Eichstadt, S.; Barriga, M.; Ponakala, A.; Teng, C.; Nguyen, N.T.; Siprashvili, Z.; Nazaroff, J.; Gorell, E.; Chiou, A.S.; Taylor, L.; et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. J. Clin. Investig. 2019, 4, e130554. [Google Scholar] [CrossRef] [Green Version]
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients with Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, V.; Choudhary, D.; Kumar Sharma, S.; Doval, D. Bone marrow transplant for recessive dystrophic epidermolysis bullosa. Pediatr. Hematol. Oncol. J. 2019, 4, 74–76. [Google Scholar] [CrossRef]
- Badiavas, E.V.; Abedi, M.; Butmarc, J.; Falanga, V.; Quesenberry, P. Participation of bone marrow derived cells in cutaneous wound healing. J. Cell Physiol. 2003, 196, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Tolar, J.; Ishida-Yamamoto, A.; Riddle, M.; McElmurry, R.T.; Osborn, M.; Xia, L.; Lund, T.; Slattery, C.; Uitto, J.; Christiano, A.M.; et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood 2009, 113, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chino, T.; Tamai, K.; Yamazaki, T.; Otsuru, S.; Kikuchi, Y.; Nimura, K.; Endo, M.; Nagai, M.; Uitto, J.; Kitajima, Y.; et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am. J. Pathol. 2008, 173, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, K.; Medina, R.J.; Kageyama, T.; Miyazaki, M.; Yoshino, T.; Makino, T.; Huh, N.-H. Participation of adult mouse bone marrow cells in reconstitution of skin. Am. J. Pathol. 2003, 163, 1227–1231. [Google Scholar] [CrossRef] [Green Version]
- Körbling, M.; Katz, R.L.; Khanna, A.; Ruifrok, A.C.; Rondon, G.; Albitar, M.; Champlin, R.E.; Estrov, Z. Hepatocytes and epithelial cells of donor origin in recipients of peripheral-blood stem cells. N. Engl. J. Med. 2002, 346, 738–746. [Google Scholar] [CrossRef]
- Tolar, J.; Wagner, J.E. Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: Repair of the extracellular matrix. Lancet 2013, 382, 1214–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, M.B.; Radhakrishnan, K.; Giller, R.; Umegaki, N.; Harel, S.; Kiuru, M.; Morel, K.D.; LeBoeuf, N.; Kandel, J.; Bruckner, A.; et al. Reduced Toxicity Conditioning and Allogeneic Hematopoietic Progenitor Cell Transplantation for Recessive Dystrophic Epidermolysis Bullosa. J. Pediatr. 2015, 167, 765–769.e1. [Google Scholar] [CrossRef]
- Gostyńska, K.; Yenamandra, V.; Lindemans, C.; Duipmans, J.; Gostyński, A.; Jonkman, M.; Boelens, J. Allogeneic Haematopoietic Cell Transplantation for Epidermolysis Bullosa: The Dutch Experience. Acta Derm. Venereol. 2019, 99, 347–348. [Google Scholar] [CrossRef] [Green Version]
- Uitto, J.; Bruckner-Tuderman, L.; McGrath, J.A.; Riedl, R.; Robinson, C. EB2017-Progress in Epidermolysis Bullosa Research toward Treatment and Cure. J. Invest. Dermatol. 2018, 138, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.; Horch, R.E.; Stachel, K.-D.; Holter, W.; Kandler, M.A.; Hertzberg, H.; Rascher, W.; Campean, V.; Carbon, R.; Schneider, H. Hematopoietic stem cell transplantation and subsequent 80% skin exchange by grafts from the same donor in a patient with Herlitz disease. Transplantation 2005, 79, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Hammersen, J.; Has, C.; Naumann-Bartsch, N.; Stachel, D.; Kiritsi, D.; Söder, S.; Tardieu, M.; Metzler, M.; Bruckner-Tuderman, L.; Schneider, H. Genotype, Clinical Course, and Therapeutic Decision Making in 76 Infants with Severe Generalized Junctional Epidermolysis Bullosa. J. Invest. Dermatol. 2016, 136, 2150–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrolia, I.; Foppiani, E.M.; Murgia, A.; Candini, O.; Samarelli, A.V.; Grisendi, G.; Veronesi, E.; Horwitz, E.M.; Dominici, M. Challenges in Clinical Development of Mesenchymal Stromal/Stem Cells: Concise Review. Stem Cells Transl. Med. 2019, 8, 1135–1148. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative Analysis of Mesenchymal Stem Cells from Bone Marrow, Umbilical Cord Blood, or Adipose Tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Bronckaers, A.; Hilkens, P.; Martens, W.; Gervois, P.; Ratajczak, J.; Struys, T.; Lambrichts, I. Mesenchymal stem/stromal cells as a pharmacological and therapeutic approach to accelerate angiogenesis. Pharmacol. Ther. 2014, 143, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Kühl, T.; Mezger, M.; Hausser, I.; Handgretinger, R.; Bruckner-Tuderman, L.; Nyström, A. High Local Concentrations of Intradermal MSCs Restore Skin Integrity and Facilitate Wound Healing in Dystrophic Epidermolysis Bullosa. Mol. Ther. 2015, 23, 1368–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conget, P.; Rodriguez, F.; Kramer, S.; Allers, C.; Simon, V.; Palisson, F.; Gonzalez, S.; Yubero, M.J. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy 2010, 12, 429–431. [Google Scholar] [CrossRef]
- Nevala-Plagemann, C.; Lee, C.; Tolar, J. Placenta-based therapies for the treatment of epidermolysis bullosa. Cytotherapy 2015, 17, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Yang, J.; Kleffel, S.; Uehara, M.; Barthel, S.R.; Schlapbach, C.; Zhan, Q.; Dudeney, S.; Mueller, H.; Lee, N.; et al. ABCB5 Identifies Immunoregulatory Dermal Cells. Cell Rep. 2015, 12, 1564–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beken, S.V.; de Vries, J.C.; Meier-Schiesser, B.; Meyer, P.; Jiang, D.; Sindrilaru, A.; Ferreira, F.F.; Hainzl, A.; Schatz, S.; Muschhammer, J.; et al. Newly Defined ATP-Binding Cassette Subfamily B Member 5 Positive Dermal Mesenchymal Stem Cells Promote Healing of Chronic Iron-Overload Wounds via Secretion of Interleukin-1 Receptor Antagonist. Stem Cells 2019, 37, 1057–1074. [Google Scholar] [CrossRef] [Green Version]
- Jayarajan, V.; Kounatidou, E.; Qasim, W.; Di, W.-L. Ex vivo gene modification therapy for genetic skin diseases—Recent advances in gene modification technologies and delivery. Exp. Dermatol. 2021, 30, 887–896. [Google Scholar] [CrossRef]
- Dellambra, E.; Pellegrini, G.; Guerra, L.; Ferrari, G.; Zambruno, G.; Mavilio, F.; De Luca, M. Toward Epidermal Stem Cell-Mediated ex Vivo Gene Therapy of Junctional Epidermolysis Bullosa. Hum. Gene Ther. 2000, 11, 2283–2287. [Google Scholar] [CrossRef]
- Rheinwatd, J.G.; Green, H. Seria cultivation of strains of human epidemal keratinocytes: The formation keratinizin colonies from single cell is. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Boyce, S.T.; Kagan, R.J.; Yakuboff, K.P.; Meyer, N.A.; Rieman, M.T.; Greenhalgh, D.G.; Warden, G.D. Cultured Skin Substitutes Reduce Donor Skin Harvesting for Closure of Excised, Full-Thickness Burns. Ann. Surg. 2002, 235, 269. [Google Scholar] [CrossRef]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; Di Nunzio, F.; Di Iorio, E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, M.C.; et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006, 12, 1397–1402. [Google Scholar] [CrossRef]
- Bauer, J.W.; Koller, J.; Murauer, E.M.; De Rosa, L.; Enzo, E.; Carulli, S.; Bondanza, S.; Recchia, A.; Muss, W.; Diem, A.; et al. Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells. J. Invest. Dermatol. 2017, 137, 778–781. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef]
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Invest. Dermatol. 2019, 139, 1221–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welponer, T.; Prodinger, C.; Pinon-Hofbauer, J.; Hintersteininger, A.; Breitenbach-Koller, H.; Bauer, J.W.; Laimer, M. Clinical Perspectives of Gene-Targeted Therapies for Epidermolysis Bullosa. Dermatol. Ther. 2021, 11, 1175–1197. [Google Scholar] [CrossRef] [PubMed]
- Araki, R.; Uda, M.; Hoki, Y.; Sunayama, M.; Nakamura, M.; Ando, S.; Sugiura, M.; Ideno, H.; Shimada, A.; Nifuji, A.; et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 2013, 494, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Umegaki-Arao, N.; Pasmooij, A.M.G.; Itoh, M.; Cerise, J.E.; Guo, Z.; Levy, B.; Gostyński, A.; Rothman, L.R.; Jonkman, M.F.; Christiano, A.M. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci. Transl. Med. 2014, 6, 264ra164. [Google Scholar] [CrossRef]
- Tolar, J.; Xia, L.; Riddle, M.J.; Lees, C.J.; Eide, C.R.; McElmurry, R.T.; Titeux, M.; Osborn, M.J.; Lund, T.C.; Hovnanian, A.; et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2011, 131, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, B.R.; Osborn, M.J.; McElroy, A.N.; Twaroski, K.; Lonetree, C.-L.; DeFeo, A.P.; Xia, L.; Eide, C.; Lees, C.J.; McElmurry, R.T.; et al. CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regen. Med. 2016, 1, 16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkuma, S.; Guo, Z.; Christiano, A.M. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc. Natl. Acad. Sci. USA 2016, 113, 5676–5681. [Google Scholar] [CrossRef] [Green Version]
- Jacków, J.; Guo, Z.; Hansen, C.; Abaci, H.E.; Doucet, Y.S.; Shin, J.U.; Hayashi, R.; DeLorenzo, D.; Kabata, Y.; Shinkuma, S.; et al. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proc. Natl. Acad. Sci. USA 2019, 116, 26846–26852. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Starker, C.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; DeFeo, A.P.; Gabriel, R.; Schmidt, M.; Von Kalle, C.; et al. TALEN-based gene correction for epidermolysis bullosa. Mol. Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Umegaki-Arao, N.; Guo, Z.; Liu, L.; Higgins, C.A.; Christiano, A.M. Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PLoS ONE 2013, 8, e77673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, M.; Kawagoe, S.; Tamai, K.; Nakagawa, H.; Asahina, A.; Okano, H.J. Footprint-free gene mutation correction in induced pluripotent stem cell (iPSC) derived from recessive dystrophic epidermolysis bullosa (RDEB) using the CRISPR/Cas9 and piggyBac transposon system. J. Dermatol. Sci. 2020, 98, 163–172. [Google Scholar] [CrossRef]
- Awe, J.P.; Lee, P.C.; Ramathal, C.; Vega-Crespo, A.; Durruthy-Durruthy, J.; Cooper, A.; Karumbayaram, S.; Lowry, W.E.; Clark, A.T.; Zack, J.A.; et al. Generation and characterization of transgene-free human induced pluripotent stem cells and conversion to putative clinical-grade status. Stem Cell Res. Ther. 2013, 4, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Classification | Cell Type | Differentiation | Examples |
|---|---|---|---|
| Totipotent | Embryonic stem cells | Differentiate into any cell type | Zygotes |
| Pluripotent | Advanced Perinatal embryonic stem cells | Differentiate into of all three germ layers | ECSs, AMSCs, CMSCs, USSCs, iPSCs, VSELs |
| Multipotent | Adult stem cells | Differentiate into a limited range of cell types | MSCs, HSCs |
| Oligopotent | Adult stem cells | Differentiate into a limited range of cell types | Lymphoid, Myeloid |
| Unipotent | Adult stem cells | Differentiate into a single type cell type | Epidermal |
| Title/Clinical Trial Identifier | Phase/Status | Condition | Interventions/Cell Types | Safety | Outcome | Country |
|---|---|---|---|---|---|---|
| NCT03529877: Allogeneic ABCB5-positive Stem Cells for Treatment of Epidermolysis Bullosa [47] | Phase I/II, International, multicentric, single-arm. Completed | RDEB | Intravenous infusion of allogeneic ABCB5+ MSCs | Good tolerability, manageable safety. Adverse effects: (1) mild lymphadenopathy (1 out of 16), (2) hypersensitivity reactions (2 out of 16). | Significant reductions in EB disease activity and scarring index activity. Reductions in pain and itch. | United States (US), Austria, France, German, Italy, United Kingdom (UK) |
| NCT04153630: Safety Study and Preliminary Efficacy of Infusion Haploidentical Mesenchymal Stem Cells Derived From Bone Marrow | Phase I/II, pilot, single group, open-label. Unknown status | RDEB | Systemic infusion of BM-MSCs from haploidentical donor. Intravenous injection with a dose of 2–3 × 106 cells/kg. | Unknown | Unknown | Spain |
| NCT04520022: Safety and Effectiveness Study of Allogeneic Umbilical Cord Blood-derived Mesenchymal Stem Cell [46] | Phase I/II, single-group, single-center, open-label. Completed. | RDEB | Intravenous administrations of allogeneic hUCB-MSCs, 3 × 106 cells/kg, total of 3 doses every 2 weeks. | Well-tolerated, no severe adverse events. | Improvements in the EB severity score, body surface area involvement, blister counts, pain, pruritus and quality of life. Increase of collagen type VII expression at the DEJ in 1 out of 6 patients. | Korea |
| NCT02579369: Study to Evaluate the Safety of ALLO-ASC-DFU in the Subjects With Dystrophic Epidermolysis Bullosa | Phase I/II, non-randomized, parallel assignment, open-label. Unknown status | RDEB | Application of hydrogel dressing for RDEB wound with allogeneic AT-MSCs | Unknown | Unknown | Korea |
| NCT00478244: Allogeneic hematopoietic cell transplantation to correct the biochemical defect and create tolerance to donor tissue in subjects with EB [50]. | Single group, open-label. Terminated (Competing studies). | RDEB | Immunomyeloablative chemotherapy and allogeneic hematopoietic stem cell transplantation | One of six patients died as a consequence of graft rejection and infection after 2 years. High-risk therapeutic approach for patients with less severe RDEB. | Increased collagen VII deposition at the DEJ in 5 of 5 recipients without normalization of anchoring fibrils and reduced blistering. | US |
| NCT03183934: A Follow-up Study to Evaluate the Efficacy and Safety of ALLO-ASC-DFU in ALLO-ASC-EB-101 Clinical Trial | Follow-up study | Application of dressing for DEB with allogeneic AT-MSCs | Unknown | Unknown | Korea | |
| NCT00881556: A pilot study of reduced intensity conditioning (RIC) and allogeneic stem cell transplantation (ALLOSCT) in children with RDEB. | Early phase I, single group, open-label. Terminated | RDEB | Reduced Intensity Conditioning (RIC) and Allogeneic Stem Cell Transplantation (AlloSCT) from family-related donors and unrelated cord blood (UCB) donors will be safe and well tolerated in selected patients with RDEB. | Unknown | Unknown | U.S. |
| NCT02582775: Biochemical Correction of Severe EB by Allo HSCT and Serial Donor MSCs [51] | Phase II, Non-randomized, open-label, Active, not recruiting | Severe, generalized RDEB | Epidermolysis bullosa patients treated with chemotherapy and BM-HSC transplant with BM-MSCs infusions | Improved safety due to PTCγ treatment. | Improved Col VII and restoration of anchoring fibrils, reduced erosions. | U.S |
| NCT01033552: Biochemical Correction of Severe EB by Allo HSCT and “Off-the-shelf” MSCs | Phase I/II, single group, open-label. Completed | Severe EB | Mesenchymal stem cell transplantation infused intravenously and bone marrow or umbilical cord blood products infusion. | Unknown | Unknow | U.S. |
| NCT02323789: Mesenchymal allogeneic stromal cells in adults with RDEB (ADSTEM) [52] | Phase I/II, Open-label, Unknown status | RDEB | Intravenous alloegeneic mesenchymal stromal cell (BM-MSCs) therapy in adults with RDEB | No serious adverse events up to 12 months. Requirement for monitoring possible development or progression of squamous cell carcinoma (SCC). | Transient reduction in disease activity scores and significant reduction in itch. Transient increase in type VII collagen at 1 out of 10 participants. | U.K. |
| 2012-00894-87: Allogeneic mesenchymal stromal cells for the treatment of skin disease in children with recessive dystrophic epidermolysis bullosa [53] | Phase I/II, Non-randomized-controlled, single arm. Completed | RDEB | 3 intravenous allogeneic BM-MSC infusions | Good tolerance | Decrease in global severity score, increase in quality of life, decrease in blister counts. | U.K. |
| NCT05111600: Open-label, Pivotal Clinical Trial to Confirm Efficacy and Safety of Autologous Grafts Containing Stem Cells Genetically Modified for Epidermis Restoration in Patients With Junctional Epidermolysis Bullosa (HOLOGENE 5) [54]. | Phase II/III, Prospective, multicenter and multinational, open-label, uncontrolled. Recruiting | JEB Non Herlitz type | Grafting of fibrin-cultured epidermal sheets generated by transgenic clonogenic keratinocytes, including epidermal stem cells | Promising safety profile | Most likely permanent functional restoration of the dermo-epidermal junction, long-lasting ie engrafted transgenic epidermal stem cells allowing continuous self-renewal | Italy, France |
| NCT02493816: Phase I Study of Lentiviral-mediated COL7A1 Gene-modified Autologous Fibroblasts in adults with Recessive Dystrophic Epidermolysis Bullosa [55]. | Phase I, open-label. Completed | RDEB | Lentiviral-mediated COL7A1 gene-modified autologous fibroblasts, 3 intra-dernal injections on day 0 only | Safe, only mild local injection procedure-related side effects lasting for a few hours without requiring treatment | Significant increase in collagen VII expression at the derm-epidermla junction but not associated with mature anchoring filaments, improvement of healing. | UK |
| NCT02810951: A Phase I/II Study of FCX-007 (Genetically-Modified Autologous Human Dermal Fibroblasts) for Recessive Dystrophic Epidermolysis Bullosa (RDEB) [56]. | Phase I/II, single group, opne-label. Terminated | RDEB | FCX-007 is a genetically modified cell product obtained from the subject’s own skin cells (Autologous fibroblasts). The cells are expanded and genetically modified to produce functional COL7. FCX-007 cell suspension is injected intradermally. | |||
| NCT03490331: Clinical trial to assess the safety and efficacy of autologous cultured epidermal grafts containing epidermal stem cells genetically modified with a gamma-retroviral (rv) vector carrying COL17A1 cDNA for restoration of epidermis in patients with junctional epidermolysis bullosa | Phase I/II, prospective, open-label, uncontrolled. Terminated (No patient ongoing (none completed the study). Changes to the viral vector ongoing) | JEB | Transplantation surgery of genetically corrected cultured epidermal autograft. | Unknown | Unknown | Austria |
| NCT02984085: Clinical trial to assess the safety and efficacy of autologous cultured epidermal grafts containing epidermal stem cells genetically modified with a gamma-retroviral (rv) vector carrying COL7A1 cDNA for restoration of epidermis in patients with recessive dystrophic epidermolysis bullosa. | Phase I/II, prospective, open-label, uncontrolled. Terminated (replaced by study in progress) | RDEB | Transplantation surgery of genetically corrected cultured epidermal autograft. | Unknown | Unknown | |
| NCT04186650: Ex vivo gene therapy linical trial for RDEB using genetically corrected autologous skin equivalents (EBGraft). | Phase I/II, non-randomized single-group, open—label. Active, not recruiting | RDEB | Graft of SIN RV-mediated COL7A1 gene-modified autologous skin equivalent | - | - | France |
| NCT01263379: Gene transfer for recessive dystophic epidrmolysis bullosa [57,58,59] | Phase I/II, single-center, open-label. Active, not recruiting. | RDEB | Gene transfer for RDEB using the drug LZRSE-Col7A1 engineered autologous epidermal sheets (EB-101) | Safe, no serious adverse effects during up to 8 years. 2 adult patients out of 10 developed SCC on anatomic sites distant form grafted sites. | Long-term improvements in wound healing, pain and itch. Some grafts showed collagen VII expression in anchoring fibrils. Collagen VII expression persisted up to 2 years after treatment in 2 participants. | US |
| NCT04227106: A phase 3 study of EB-101 for the treatment of RDEB. | Phase III, single-group, open-label. Completed | RDEB | One-time surgical application of EB-101 on up to 6 chronic RDEB wounds. | Unknown | Unknown | US |
| NCT051116000: Open-label, pivotal clinical trial to confirm efficacy and safety of autologous grafts containing stem cells genetically modified for epidermis restoration in patients with JEB (HOLOGENE 5). | Phase II/III, prospective, multicenter and multinational, open-label, uncontrolled. | JEB | Transplantation of autologous cultured epidermal grafts containing epidermal stem cells genetically modified transduced with a LAMB3-gamma retroviral vector. | In progress | In progress | France, Italy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niti, A.; Koliakos, G.; Michopoulou, A. Stem Cell Therapies for Epidermolysis Bullosa Treatment. Bioengineering 2023, 10, 422. https://doi.org/10.3390/bioengineering10040422
Niti A, Koliakos G, Michopoulou A. Stem Cell Therapies for Epidermolysis Bullosa Treatment. Bioengineering. 2023; 10(4):422. https://doi.org/10.3390/bioengineering10040422
Chicago/Turabian StyleNiti, Argyrw, Georgios Koliakos, and Anna Michopoulou. 2023. "Stem Cell Therapies for Epidermolysis Bullosa Treatment" Bioengineering 10, no. 4: 422. https://doi.org/10.3390/bioengineering10040422
APA StyleNiti, A., Koliakos, G., & Michopoulou, A. (2023). Stem Cell Therapies for Epidermolysis Bullosa Treatment. Bioengineering, 10(4), 422. https://doi.org/10.3390/bioengineering10040422