




Towards a Rapid-Turnaround Low-Depth Unbiased Metagenomics Sequencing Workflow on the Illumina Platforms

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
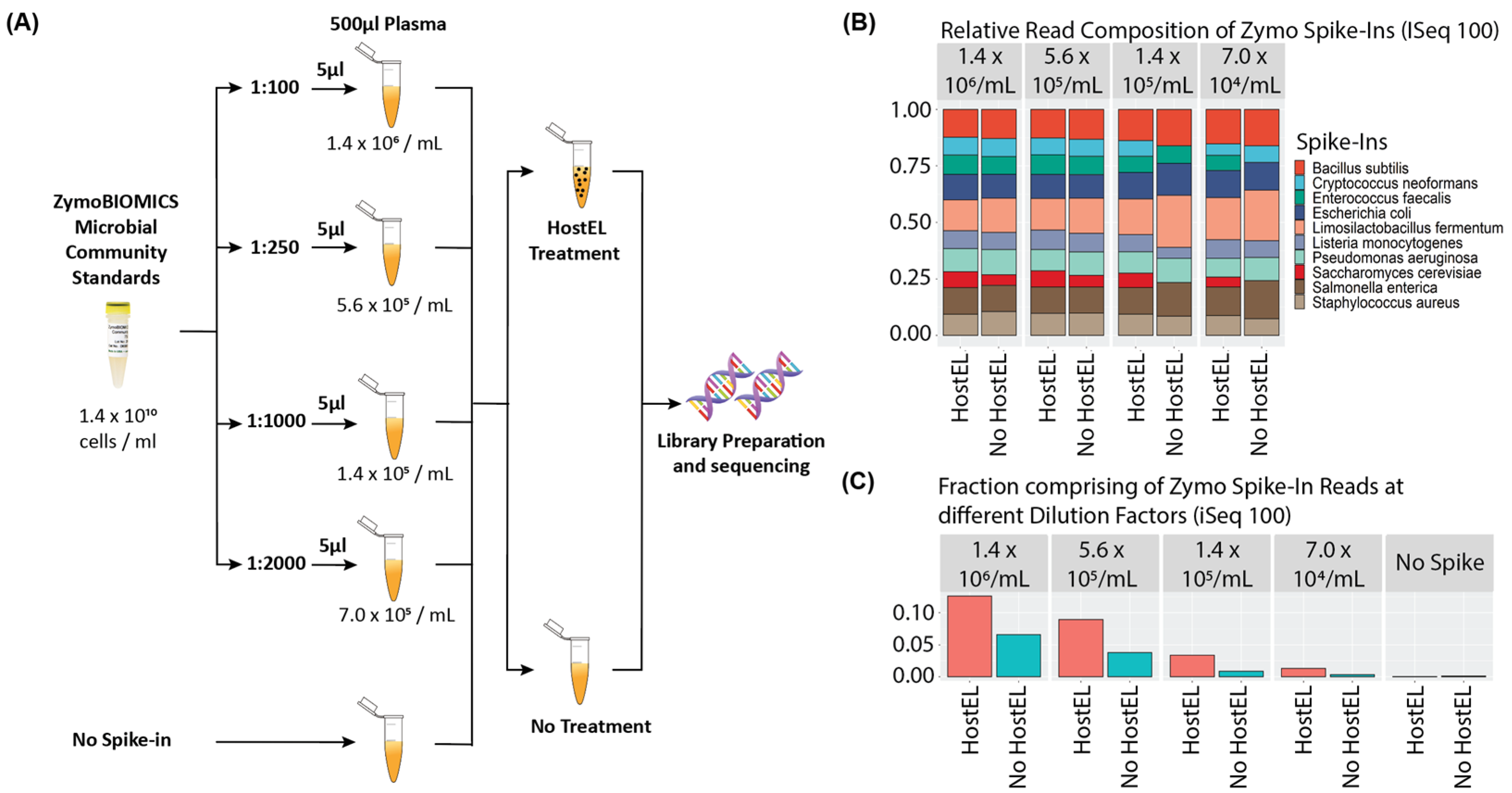
2.1. Analytical Validation
2.2. Sequencing on iSeq 100 System and MiniSeq System
2.3. Data Analysis
2.4. Sample Collection and Ethics Statement
2.5. qPCR Validation
3. Results
3.1. Principles of HostEL and AmpRE
3.2. Low Depth Sequencing Can Detect Physiological Levels of Bacteria
3.3. HostEL Enhances Sensitivity and Specificity of Low Depth Sequencing
3.4. Concordance with Banked Patient Plasma Samples Is >90%
3.5. Both DNA and RNA Viruses Can Be Detected If Ct Is Less than 33
3.6. Rapid Turnaround Can Be Achieved with Different Sequencing Platforms
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armitage, C. The High Burden of Infectious Disease. Nature 2021, 598, S9. [Google Scholar] [CrossRef]
- Yang, S.; Rothman, R.E. PCR-Based Diagnostics for Infectious Diseases: Uses, Limitations, and Future Applications in Acute-Care Settings. Lancet Infect. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef]
- Jia, X.; Hu, L.; Wu, M.; Ling, Y.; Wang, W.; Lu, H.; Yuan, Z.; Yi, Z.; Zhang, X. A Streamlined Clinical Metagenomic Sequencing Protocol for Rapid Pathogen Identification. Sci. Rep. 2021, 11, 4405. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Chiu, C. The Role of Metagenomics and Next-Generation Sequencing in Infectious Disease Diagnosis. Clin. Chem. 2022, 68, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Hong, D.K.; Blauwkamp, T.A.; Kertesz, M.; Bercovici, S.; Truong, C.; Banaei, N. Liquid Biopsy for Infectious Diseases: Sequencing of Cell-Free Plasma to Detect Pathogen DNA in Patients with Invasive Fungal Disease. Diagn. Microbiol. Infect. Dis. 2018, 92, 210–213. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory Validation of a Clinical Metagenomic Sequencing Assay for Pathogen Detection in Cerebrospinal Fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced Methods for Unbiased Deep Sequencing of Lassa and Ebola RNA Viruses from Clinical and Biological Samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, S.; Carroll, K.C. Understanding the Promises and Hurdles of Metagenomic Next-Generation Sequencing as a Diagnostic Tool for Infectious Diseases. Clin. Infect. Dis. 2018, 66, 778–788. [Google Scholar] [CrossRef]
- Morales, M. The Next Big Thing? Next-Generation Sequencing of Microbial Cell-Free DNA Using the Karius Test. Clin. Microbiol. Newsl. 2021, 43, 69–79. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Chiu, C.Y. Viral Pathogen Discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Breitwieser, F.P.; Kumar, A.; Hao, H.; Burger, P.; Rodriguez, F.J.; Lim, M.; Quiñones-Hinojosa, A.; Gallia, G.L.; Tornheim, J.A.; et al. Next-Generation Sequencing in Neuropathologic Diagnosis of Infections of the Nervous System. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e251. [Google Scholar] [CrossRef]
- Cazanave, C.; Greenwood-Quaintance, K.E.; Hanssen, A.D.; Karau, M.J.; Schmidt, S.M.; Gomez Urena, E.O.; Mandrekar, J.N.; Osmon, D.R.; Lough, L.E.; Pritt, B.S.; et al. Rapid Molecular Microbiologic Diagnosis of Prosthetic Joint Infection. J. Clin. Microbiol. 2013, 51, 2280–2287. [Google Scholar] [CrossRef]
- Naccache, S.N.; Peggs, K.S.; Mattes, F.M.; Phadke, R.; Garson, J.A.; Grant, P.; Samayoa, E.; Federman, S.; Miller, S.; Lunn, M.P.; et al. Diagnosis of Neuroinvasive Astrovirus Infection in an Immunocompromised Adult with Encephalitis by Unbiased Next-Generation Sequencing. Clin. Infect. Dis. 2015, 60, 919–923. [Google Scholar] [CrossRef]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable Diagnosis of Neuroleptospirosis by Next-Generation Sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef]
- Heravi, F.S.; Zakrzewski, M.; Vickery, K.; Hu, H. Host DNA Depletion Efficiency of Microbiome DNA Enrichment Methods in Infected Tissue Samples. J. Microbiol. Methods 2020, 170, 105856. [Google Scholar] [CrossRef]
- Gu, W.; Deng, X.; Lee, M.; Sucu, Y.D.; Arevalo, S.; Stryke, D.; Federman, S.; Gopez, A.; Reyes, K.; Zorn, K.; et al. Rapid Pathogen Detection by Metagenomic Next-Generation Sequencing of Infected Body Fluids. Nat. Med. 2021, 27, 115–124. [Google Scholar] [CrossRef]
- Thoendel, M.; Jeraldo, P.R.; Greenwood-Quaintance, K.E.; Yao, J.Z.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Comparison of Microbial DNA Enrichment Tools for Metagenomic Whole Genome Sequencing. J. Microbiol. Methods 2016, 127, 141–145. [Google Scholar] [CrossRef]
- Darton, T.; Guiver, M.; Naylor, S.; Jack, D.L.; Kaczmarski, E.B.; Borrow, R.; Read, R.C. Severity of Meningococcal Disease Associated with Genomic Bacterial Load. Clin. Infect. Dis. 2009, 48, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.R.; Rawat, A.; Tang, P.; Jithesh, P.V.; Thomas, E.; Tan, R.; Tilley, P. Depletion of Human DNA in Spiked Clinical Specimens for Improvement of Sensitivity of Pathogen Detection by Next-Generation Sequencing. J. Clin. Microbiol. 2016, 54, 919–927. [Google Scholar] [CrossRef]
- van Boheemen, S.; van Rijn, A.L.; Pappas, N.; Carbo, E.C.; Vorderman, R.H.; Sidorov, I.; vant Hof, P.J.; Mei, H.; Claas, E.C.; Kroes, A.C.; et al. Retrospective Validation of a Metagenomic Sequencing Protocol for Combined Detection of RNA and DNA Viruses Using Respiratory Samples from Pediatric Patients. J. Mol. Diagn. 2020, 22, 196–207. [Google Scholar] [CrossRef]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore Sequencing of Influenza Virus Direct from Clinical Respiratory Samples. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef]
- Zinter, M.S.; Dvorak, C.C.; Mayday, M.Y.; Iwanaga, K.; Ly, N.P.; McGarry, M.E.; Church, G.D.; Faricy, L.E.; Rowan, C.M.; Hume, J.R.; et al. Pulmonary Metagenomic Sequencing Suggests Missed Infections in Immunocompromised Children. Clin. Infect. Dis. 2019, 68, 1847–1855. [Google Scholar] [CrossRef]
- Allicock, O.M.; Guo, C.; Uhlemann, A.-C.; Whittier, S.; Chauhan, L.V.; Garcia, J.; Price, A.; Morse, S.S.; Mishra, N.; Briese, T.; et al. BacCapSeq: A Platform for Diagnosis and Characterization of Bacterial Infections. mBio 2018, 9, e02007-18. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic Sequencing with Spiked Primer Enrichment for Viral Diagnostics and Genomic Surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef]
- Petty, T.J.; Cordey, S.; Padioleau, I.; Docquier, M.; Turin, L.; Preynat-Seauve, O.; Zdobnov, E.M.; Kaiser, L. Comprehensive Human Virus Screening Using High-Throughput Sequencing with a User-Friendly Representation of Bioinformatics Analysis: A Pilot Study. J. Clin. Microbiol. 2014, 52, 3351–3361. [Google Scholar] [CrossRef]
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A.; et al. Capturing Sequence Diversity in Metagenomes with Comprehensive and Scalable Probe Design. Nat. Biotechnol. 2019, 37, 160–168. [Google Scholar] [CrossRef]
- Nelson, M.T.; Pope, C.E.; Marsh, R.L.; Wolter, D.J.; Weiss, E.J.; Hager, K.R.; Vo, A.T.; Brittnacher, M.J.; Radey, M.C.; Hayden, H.S.; et al. Human and Extracellular DNA Depletion for Metagenomic Analysis of Complex Clinical Infection Samples Yields Optimized Viable Microbiome Profiles. Cell Rep. 2019, 26, 2227–2240.e5. [Google Scholar] [CrossRef]
- Metagenomic Sequencing for Infectious Diseases Diagnostics with Charles Chiu. Available online: https://asm.org:443/Podcasts/MTM/Episodes/Metagenomic-Sequencing-for-Infectious-Diseases-Dia (accessed on 17 March 2023).
- ZymoBIOMICS™ Microbial Community Standard (D6300). Available online: https://files.zymoresearch.com/protocols/_d6300_zymobiomics_microbial_community_standard.pdf (accessed on 29 March 2022).
- Pinto, G.G.; Poloni, J.A.T.; Paskulin, D.D.; Spuldaro, F.; Paris, F.; Barth, A.L.; Manfro, R.C.; Keitel, E.; Pasqualotto, A.C. Quantitative detection of BK virus in kidney transplant recipients: A prospective validation study. J. Bras. Nefrol. 2018, 40, 59–65. [Google Scholar] [CrossRef]
- Alm, E.; Lesko, B.; Lindegren, G.; Ahlm, C.; Söderholm, S.; Falk, K.I.; Lagerqvist, N. Universal single-probe RT-PCR assay for diagnosis of dengue virus infections. PLoS Negl. Trop. Dis. 2014, 8, e3416. [Google Scholar] [CrossRef]
- Liu, C.; Chang, L.; Jia, T.; Guo, F.; Zhang, L.; Ji, H.; Zhao, J.; Wang, L. Real-time PCR assays for hepatitis B virus DNA quantification may require two different targets. Virol. J. 2017, 14, 94. [Google Scholar] [CrossRef]
- Warkad, S.D.; Nimse, S.B.; Song, K.S.; Kim, T. Development of a Method for Screening and Genotyping of HCV 1a, 1b, 2, 3, 4, and 6 Genotypes. ACS Omega 2020, 5, 10794–10799. [Google Scholar] [CrossRef]
- Germer, J.J.; Ankoudinova, I.; Belousov, Y.S.; Mahoney, W.; Dong, C.; Meng, J.; Mandrekar, J.N.; Yao, J.D. Hepatitis E Virus (HEV) Detection and Quantification by a Real-Time Reverse Transcription-PCR Assay Calibrated to the World Health Organization Standard for HEV RNA. J. Clin. Microbiol. 2017, 55, 1478–1487. [Google Scholar] [CrossRef]
- Harbecke, R.; Oxman, M.N.; Arnold, B.A.; Ip, C.; Johnson, G.R.; Levin, M.J.; Gelb, L.D.; Schmader, K.E.; Straus, S.E.; Wang, H.; et al. A real-time PCR assay to identify and discriminate among wild-type and vaccine strains of varicella-zoster virus and herpes simplex virus in clinical specimens, and comparison with the clinical diagnoses. J. Med. Virol. 2009, 81, 1310–1322. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clinical Diagnosis | iSeq 100 Detection | iSeq 100 Count | MiniSeq Detection | MiniSeq Count | qPCR Ct |
|---|---|---|---|---|---|---|
| B012 | Hepatitis C Virus | Hepatitis C Virus | 29 | Hepatitis C Virus | 72 | 23.8 |
| B013 | Hepatitis C Virus | Hepatitis C Virus | 32 | Hepatitis C Virus | 66 | 27.2 |
| B014 | HIV-1 | HIV-1 | 10 | HIV-1 | 26 | - |
| B015 | HIV-1 | HIV-1 | 2 | HIV-1 | 3 | - |
| B016 | Hepatitis B Virus | Hepatitis B virus | 4238 | Hepatitis B virus | 11,394 | 17.9 |
| B017 | Hepatitis B Virus | Hepatitis B virus | 495 | Hepatitis B virus | 1226 | 24.4 |
| B018 | Hepatitis E Virus | - | 0 | - | 0 | 34.3 |
| B019 | Hepatitis E Virus | - | 0 | - | 0 | 37.1 |
| B020 | Cytomegalovirus | Cytomegalovirus | 9 | Cytomegalovirus | 10 | - |
| B021 | Cytomegalovirus | Cytomegalovirus | 22 | Cytomegalovirus | 100 | - |
| B022 | Varicella Zoster | - | 0 | - | 0 | 33.0 |
| B023 | BK virus | - | 0 | - | 0 | 32.0 |
| B024 | BK virus | - | 0 | - | 0 | 33.0 |
| B025 | Epstein–Barr virus | Epstein–Barr virus | 13 | Epstein–Barr virus | 2 | - |
| B040 | Dengue type 4 | - | 0 | - | 0 | Neg |
| B041 | Dengue type 2 | Dengue virus | 2 | Dengue virus | 3 | - |
| B042 | Dengue type 1 | - | 0 | - | 0 | Neg |
| B043 | Dengue type 3 | Dengue virus | 7 | Dengue virus | 18 | - |
| B045 | Hepatitis B Virus | Hepatitis B virus | 7427 | Hepatitis B virus | 23,336 | 22.1 |
| B046 | Hepatitis B Virus | - | 0 | - | 0 | Neg |
| B047 | Hepatitis B Virus | - | 0 | - | 0 | 43.8 |
| B048 | Hepatitis B Virus | Hepatitis B virus | 2 | Hepatitis B virus | 2 | 38.6 |
| B049 | Hepatitis B Virus | Hepatitis B virus | 118 | Hepatitis B virus | 318 | 29.5 |
| B050 | Hepatitis B Virus | - | 0 | - | 0 | Neg |
| B051 | Hepatitis C Virus | Hepatitis C Virus | 17 | Hepatitis C Virus | 128 | 23.3 |
| B052 | Hepatitis C Virus | Hepatitis C Virus | 6 | Hepatitis C Virus | 8 | 26.7 |
| B053 | Hepatitis C Virus | Hepatitis C Virus | 2 | Hepatitis C Virus | 20 | 24.8 |
| B054 | Hepatitis C Virus | - | 0 | - | 0 | 35.0 |
| B056 | Hepatitis C Virus | Hepatitis C Virus | 4 | - | 0 | 32.9 |
| B057 | Hepatitis C Virus | - | 0 | - | 0 | 35.9 |
| B058 | Hepatitis C Virus | - | 0 | - | 0 | 34.1 |
| B059 | Hepatitis C Virus | Hepatitis C Virus | 7 | Hepatitis C Virus | 2 | 28.8 |
| B060 | Hepatitis C Virus | Hepatitis C Virus | 8 | Hepatitis C Virus | 2 | 27.9 |
| B061 | Hepatitis C Virus | - | 0 | - | 0 | 30.1 |
| B068 | No Infection | - | 0 | - | 0 | - |
| B069 | No Infection | - | 0 | - | 0 | - |
| B070 | No Infection | - | 0 | - | 0 | - |
| B071 | No Infection | - | 0 | - | 0 | - |
| B072 | No Infection | - | 0 | - | 0 | - |
| B073 | No Infection | - | 0 | - | 0 | - |
| B074 | No Infection | - | 0 | - | 0 | - |
| B075 | No Infection | - | 0 | - | 0 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koh, W.L.C.; Poh, S.E.; Lee, C.K.; Chan, T.H.M.; Yan, G.; Kong, K.W.; Lau, L.; Lee, W.Y.T.; Cheng, C.; Hoon, S.; et al. Towards a Rapid-Turnaround Low-Depth Unbiased Metagenomics Sequencing Workflow on the Illumina Platforms. Bioengineering 2023, 10, 520. https://doi.org/10.3390/bioengineering10050520
Koh WLC, Poh SE, Lee CK, Chan THM, Yan G, Kong KW, Lau L, Lee WYT, Cheng C, Hoon S, et al. Towards a Rapid-Turnaround Low-Depth Unbiased Metagenomics Sequencing Workflow on the Illumina Platforms. Bioengineering. 2023; 10(5):520. https://doi.org/10.3390/bioengineering10050520
Chicago/Turabian StyleKoh, Winston Lian Chye, Si En Poh, Chun Kiat Lee, Tim Hon Man Chan, Gabriel Yan, Kiat Whye Kong, Lalita Lau, Wai Yip Thomas Lee, Clark Cheng, Shawn Hoon, and et al. 2023. "Towards a Rapid-Turnaround Low-Depth Unbiased Metagenomics Sequencing Workflow on the Illumina Platforms" Bioengineering 10, no. 5: 520. https://doi.org/10.3390/bioengineering10050520
APA StyleKoh, W. L. C., Poh, S. E., Lee, C. K., Chan, T. H. M., Yan, G., Kong, K. W., Lau, L., Lee, W. Y. T., Cheng, C., Hoon, S., & Seow, Y. (2023). Towards a Rapid-Turnaround Low-Depth Unbiased Metagenomics Sequencing Workflow on the Illumina Platforms. Bioengineering, 10(5), 520. https://doi.org/10.3390/bioengineering10050520