
High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases




Abstract
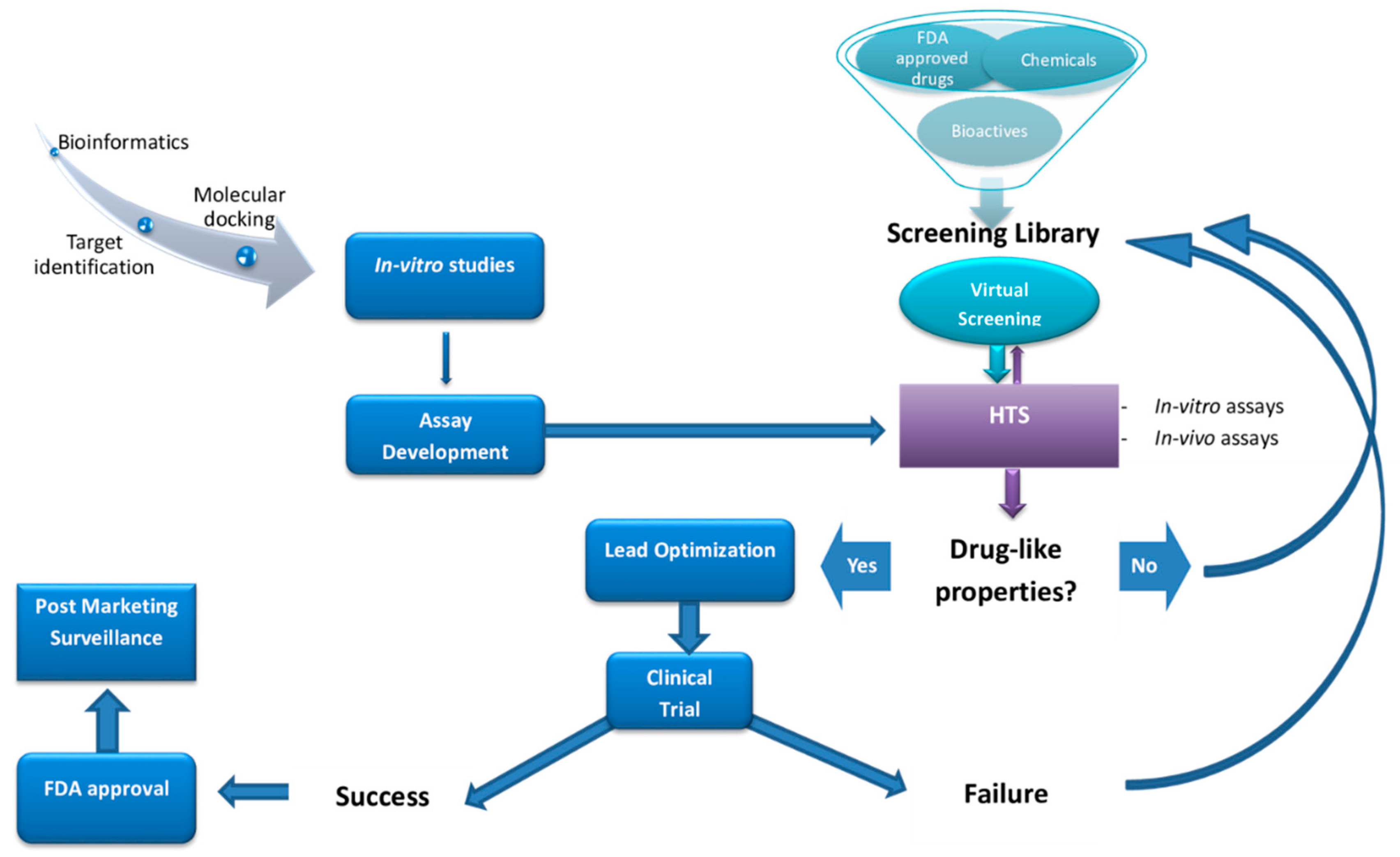
:1. Introduction
2. High-Throughput Screening (HTS)
2.1. Formats and Major Considerations for HTS Platforms
2.2. Main Types of HTS Assays
2.2.1. Cell-Based Assays
2.2.2. Biochemical Assays
2.3. Economics of HTS
3. Drugs Discovery for NDDs
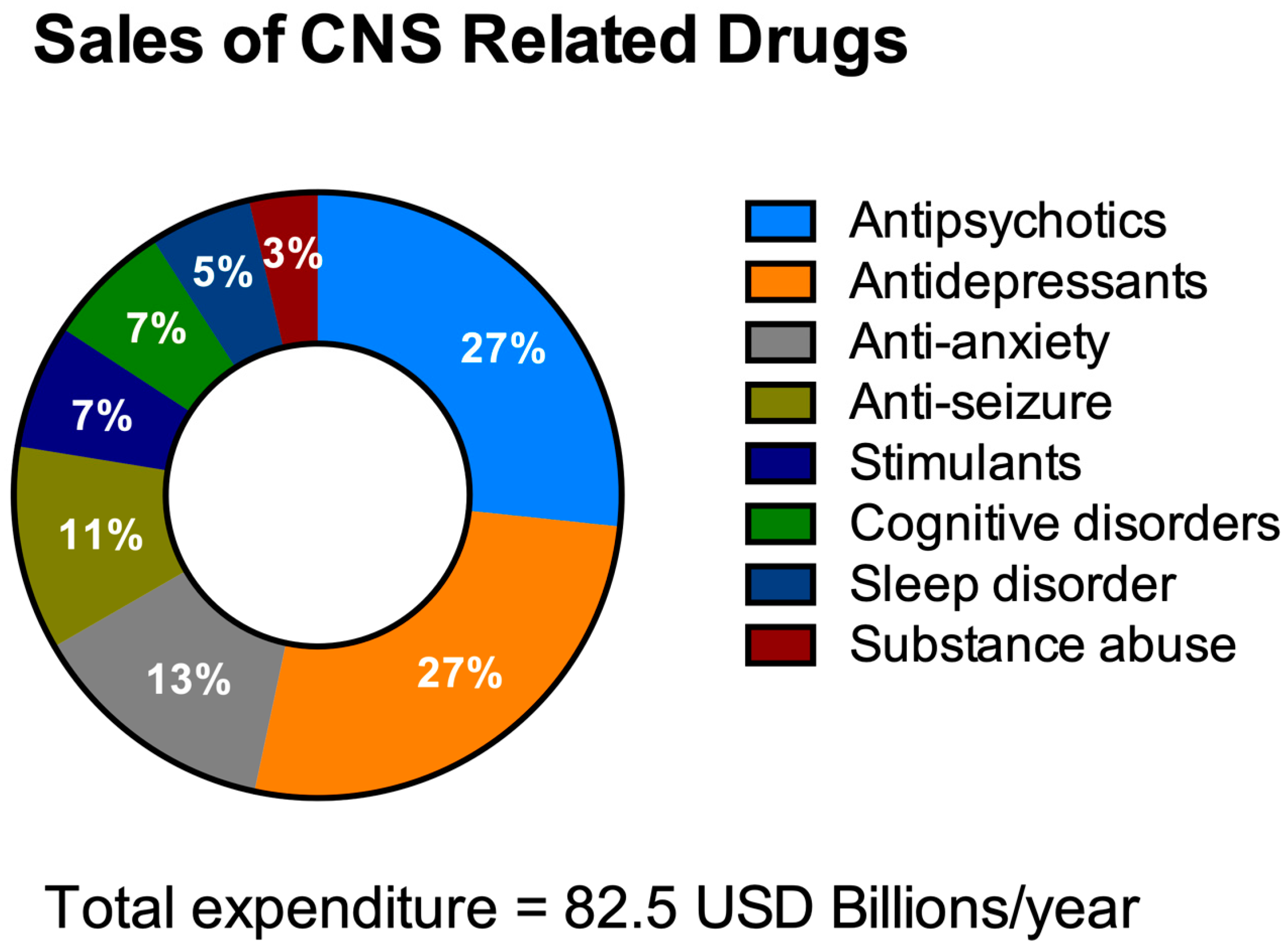
3.1. Challenges in the Discovery of CNS Drugs
3.2. The Need for HTS in the Discovery of Drugs for NDDs
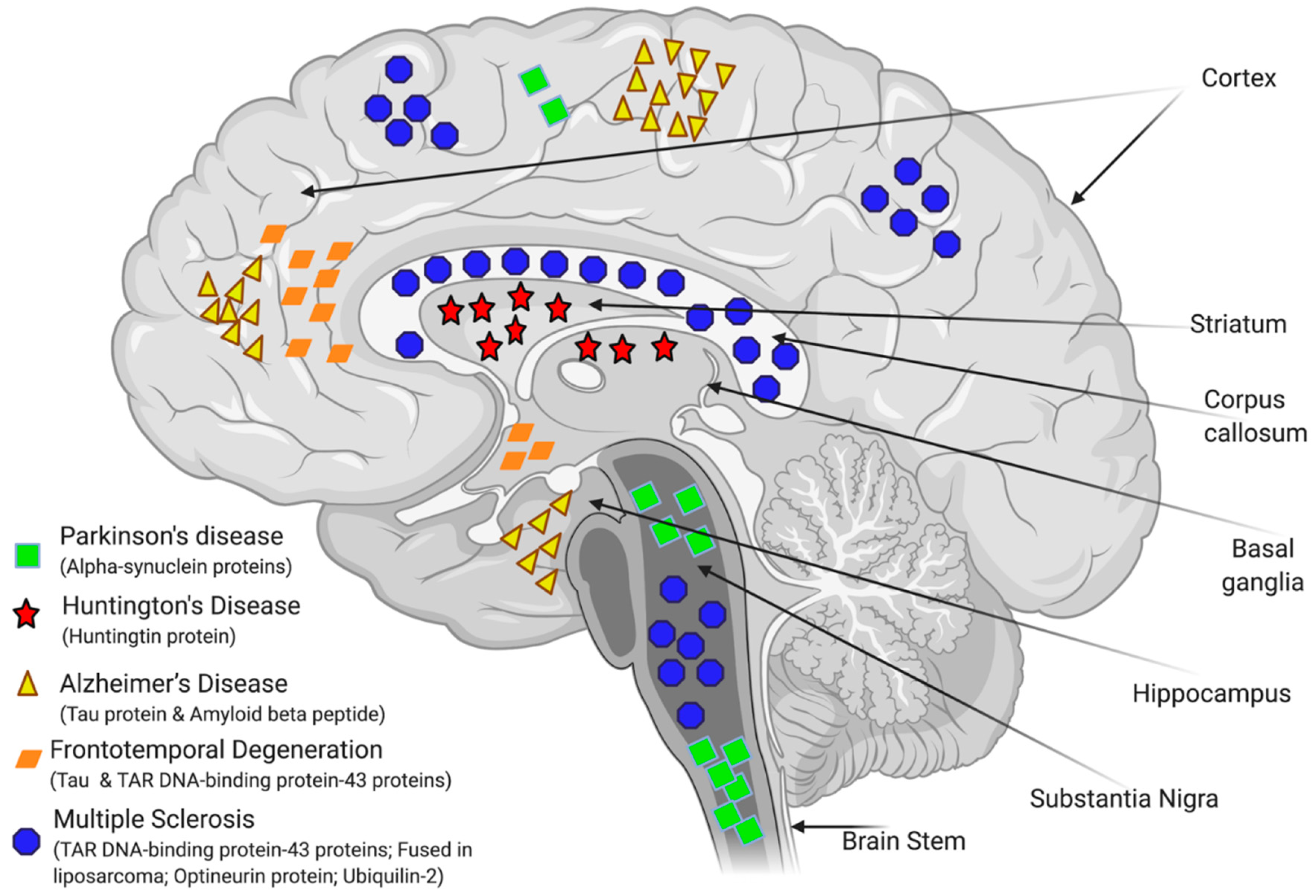
3.3. Modelling of NDDs for HTS
4. Current Challenges and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A. Strategies for discovering drugs from previously unexplored natural products. Drug Discov. Today 2000, 5, 294–300. [Google Scholar] [CrossRef]
- Al-Ali, H. The evolution of drug discovery: From phenotypes to targets, and back. MedChemComm 2016, 7, 788–798. [Google Scholar] [CrossRef]
- Johnson, E.O.; Hung, D.T. A Point of Inflection and Reflection on Systems Chemical Biology. ACS Chem. Biol. 2019, 14, 2497–2511. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.G.; Pratt, M.R. Click Chemistry in Proteomic Investigations. Cell 2020, 180, 605–632. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Zalewska, A.; Gerreth, A.K. Salivary Redox Biomarkers in Selected Neurodegenerative Diseases. J. Clin. Med. 2020, 9, 497. [Google Scholar] [CrossRef] [Green Version]
- Rabanel, J.-M.; Perrotte, M.; Ramassamy, C. Nanotechnology at the Rescue of Neurodegenerative Diseases: Tools for Early Diagnostic. In Nanobiotechnology in Neurodegenerative Diseases; Springer International Publishing: New York, NY, USA, 2019; pp. 19–48. [Google Scholar]
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current Therapeutic Molecules and Targets in Neurodegenerative Diseases Based on in silico Drug Design. Curr. Neuropharmacol. 2018, 16, 649–663. [Google Scholar] [CrossRef]
- Varma, H.; Lo, D.C.; Stockwell, B.R. High Throughput Screening for Neurodegeneration and Complex Disease Phenotypes. Comb. Chem. High Throughput Screen. 2008, 11, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Seidenbecher, C.I. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef]
- Galluzzi, L.; Joza, N.; Tasdemir, E.; Maiuri, M.C.; Hengartner, M.O.; Abrams, J.M.; Tavernarakis, N.; Penninger, J.M.; Madeo, F.; Kroemer, G. No death without life: Vital functions of apoptotic effectors. Cell Death Differ. 2008, 15, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Daub, A.; Sharma, P.; Finkbeiner, S. High-content screening of primary neurons: Ready for prime time. Curr. Opin. Neurobiol. 2009, 19, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Ando, D.; Daub, A.; Kaye, J.A.; Finkbeiner, S. High-Throughput Screening in Primary Neurons. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 506, pp. 331–360. [Google Scholar]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Okano, H. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nat. Cell Biol. 2006, 443, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Kanai, K.; Shibuya, K.; Sato, Y.; Misawa, S.; Nasu, S.; Sekiguchi, Y.; Mitsuma, S.; Isose, S.; Fujimaki, Y.; Ohmori, S.; et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2012, 83, 734–738. [Google Scholar] [CrossRef] [Green Version]
- Kanai, K.; Kuwabara, S.; Misawa, S.; Tamura, N.; Ogawara, K.; Nakata, M.; Sawai, S.; Hattori, T.; Bostock, H. Altered axonal excitability properties in amyotrophic lateral sclerosis: Impaired potassium channel function related to disease stage. Brain 2006, 129, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Tang, I.C.; Wang, J.; Yang, S.T. Cell-based assays in high-throughput screening for drug discovery. Int. J. Biotechnol. Wellness Ind. 2012, 1, 31–51. [Google Scholar]
- Eggeling, C.; Brand, L.; Ullmann, D.; Jäger, S. Highly sensitive fluorescence detection technology currently available for HTS. Drug Discov. Today 2003, 8, 632–641. [Google Scholar] [CrossRef]
- An, W.F.; Tolliday, N.J. Introduction: Cell-Based Assays for High-Throughput Screening; Springer International Publishing: New York, NY, USA, 2009; pp. 1–12. [Google Scholar]
- Kaminski, T.; Geschwindner, S. Perspectives on optical biosensor utility in small-molecule screening. Expert Opin. Drug Discov. 2017, 12, 1083–1086. [Google Scholar] [CrossRef]
- Kaminski, T.; Gunnarsson, A.; Geschwindner, S. Harnessing the Versatility of Optical Biosensors for Target-Based Small-Molecule Drug Discovery. ACS Sens. 2016, 2, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Macarrón, R.; Hertzberg, R.P. Design and Implementation of High Throughput Screening Assays. Mol. Biotechnol. 2010, 47, 270–285. [Google Scholar] [CrossRef]
- El Harrad, L.; Bourais, I.; Mohammadi, H.; Amine, A. Recent Advances in Electrochemical Biosensors Based on Enzyme Inhibition for Clinical and Pharmaceutical Applications. Sensors 2018, 18, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlarek, D.; Vorobii, M.; Ogieglo, W.; Knoll, W.; Rodriguez-Emmenegger, C.; Dostálek, J. Compact Grating-Coupled Biosensor for the Analysis of Thrombin. ACS Sens. 2019, 4, 2109–2116. [Google Scholar] [CrossRef]
- Pourbasheer, E.; Ganjali, M.R. Recent Advances in Biosensors Based Nanostructure for Pharmaceutical Analysis. Curr. Anal. Chem. 2019, 15, 152–158. [Google Scholar] [CrossRef]
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef] [Green Version]
- Kelley, B.P.; Lunn, M.R.; Root, D.E.; Flaherty, S.P.; Martino, A.M.; Stockwell, B.R. A Flexible Data Analysis Tool for Chemical Genetic Screens. Chem. Biol. 2004, 11, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Varma, H.; Lo, D.C.; Stockwell, B.R. High-Throughput and High-Content Screening for Huntington’s Disease Therapeutics. In Neurobiology of Huntington’s Disease: Applications to Drug Discovery; Lo, D.C., Hughes, R.E., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 121–1461. [Google Scholar]
- Cader, Z.; Graf, M.; Burcin, M.; Mandenius, C.-F.; Ross, J.A. Cell-Based Assays Using Differentiated Human Induced Pluripotent Cells; Springer International Publishing: New York, NY, USA, 2019; Volume 1994, pp. 1–14. [Google Scholar]
- Mandenius, C.-F.; Ross, J.A. Cell-Based Assays Using IPSCs for Drug Development and Testing; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Lee, D.W.; Doh, I.; Nam, D.-H. Unified 2D and 3D cell-based high-throughput screening platform using a micropillar/microwell chip. Sens. Actuators B Chem. 2016, 228, 523–528. [Google Scholar] [CrossRef]
- Kelm, J.M.; Lal-Nag, M.; Sittampalam, G.S.; Ferrer, M. Translational in vitro research: Integrating 3D drug discovery and development processes into the drug development pipeline. Drug Discov. Today 2019, 24, 26–30. [Google Scholar] [CrossRef]
- Wildey, M.J.; Boyd, H.; Dale, I.L.; Dahl, G.; Nicolaus, F.; Bowen, W.; Lindmark, H. Chapter Five—High-Throughput Screening, in Annual Reports in Medicinal Chemistry; Goodnow, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 149–195. [Google Scholar]
- Fu, J.; Na, Z.; Peng, B.; Uttamchandani, M.; Yao, S.Q. Accelerated cellular on- and off-target screening of bioactive compounds using microarrays. Org. Biomol. Chem. 2015, 14, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Nierode, G.; Kwon, P.S.; Dordick, J.S.; Kwon, S.-J. Cell-Based Assay Design for High-Content Screening of Drug Candidates. J. Microbiol. Biotechnol. 2016, 26, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Rue, S.M.; Anderson, P.W.; Gaylord, M.R.; Miller, J.J.; Glaser, S.M.; Lesley, S.A. A High-Throughput System for Transient and Stable Protein Production in Mammalian Cells. In Methods in Molecular Biology; Springer International Publishing: New York, NY, USA, 2019; Volume 2025, pp. 93–142. [Google Scholar]
- Damavandi, N.; Raigani, M.; Joudaki, A.; Davami, F.; Zeinali, S. Rapid characterization of the CHO platform cell line and identification of pseudo attP sites for PhiC31 integrase. Protein Expr. Purif. 2017, 140, 60–64. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Taylor, L.H.J.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and Molecular Mechanisms of the Rapid Tonicity-induced Relocalization of the Aquaporin 4 Channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)-and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; Macdonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta BBA Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Wyler, M.R.; Smith, D.H.; Cayanis, E.; Többen, U.; Aulner, N.; Mayer, T. Cell-Based Assays to Probe the ERK MAP Kinase Pathway in Endothelial Cells. In Advanced Structural Safety Studies; Springer International Publishing: New York, NY, USA, 2009; Volume 486, pp. 29–41. [Google Scholar]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Current status on Alzheimer disease molecular genetics: From past, to present, to future. Hum. Mol. Genet. 2010, 19, R4–R11. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R.; Bandmann, O. Parkinson’s disease: Insights from pathways. Hum. Mol. Genet. 2010, 19, R21–R27. [Google Scholar] [CrossRef] [Green Version]
- Haggarty, S.J.; Silva, M.C.; Cross, A.; Brandon, N.J.; Perlis, R.H. Advancing drug discovery for neuropsychiatric disorders using patient-specific stem cell models. Mol. Cell. Neurosci. 2016, 73, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, F.; Kessler, T.; Weindl, D.; Shadrin, A.; Schmiester, L.; Hache, H.; Muradyan, A.; Schütte, M.; Lim, J.-H.; Heinig, M.; et al. Efficient Parameter Estimation Enables the Prediction of Drug Response Using a Mechanistic Pan-Cancer Pathway Model. Cell Syst. 2018, 7, 567–579.e6. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y. Ligand–receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 2012, 7, 969–988. [Google Scholar] [CrossRef]
- Stoddart, L.A.; White, C.W.; Nguyen, K.; Hill, S.J.; Pfleger, K.D. Fluorescence-and bioluminescence-based approaches to study GPCR ligand binding. Br. J. Pharmacol. 2016, 173, 3028–3037. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Romano, M.; Baralle, F.E. TDP-43 high throughput screening analyses in neurodegeneration: Advantages and pitfalls. Mol. Cell. Neurosci. 2013, 56, 465–474. [Google Scholar] [CrossRef]
- Ballatore, C.; Brunden, K.; Crowe, A.; Huryn, D.; Lee, V.; Trojanowski, J.; Smith, A.; Huang, R.; Huang, W.; Johnson, R.; et al. Aminothienopyridazine Inhibitors of Tau Assembly. Patent WO2011037985 A8, 31 March 2011. [Google Scholar]
- Brunden, K.R.; Ballatore, C.; Crowe, A.; Smith, A.B., III; Lee, V.M.Y.; Trojanowski, J.Q. Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: A focus on tau assembly inhibitors. Exp. Neurol. 2010, 223, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mello, C.P.P.; Rumsey, J.; Slaughter, V.; Hickman, J.J. A human-on-a-chip approach to tackling rare diseases. Drug Discov. Today 2019, 24, 2139–2151. [Google Scholar] [CrossRef]
- Khan, N.I.; Song, E. Lab-on-a-Chip Systems for Aptamer-Based Biosensing. Micromachines 2020, 11, 220. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- MaCarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Pereira, A.D.; Williams, A.J. Origin and evolution of high throughput screening. Br. J. Pharmacol. 2007, 152, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, J.; Gustavsson, A.; Svensson, M.Y.; Wittchen, H.; Jonsson, B.H.; on behalf of the CDBE2010 Study Group; Council, T.E.B. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2011, 19, 155–162. [Google Scholar] [CrossRef]
- Nutt, D.J. The full cost and burden of disorders of the brain in Europe exposed for the first time. Eur. Neuropsychopharmacol. 2011, 21, 715–717. [Google Scholar] [CrossRef]
- Pardridge, W.M. Why is the global CNS pharmaceutical market so under-penetrated? Drug Discov. Today 2002, 7, 5–7. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; De Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Biogen. 221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT02484547 (accessed on 23 February 2021).
- Bloomberg. Biogen to Spend $2.5 Billion Before Alzheimer’s Drug Results, in 2015. Available online: https://www.bloomberg.com/news/articles/2015-04-27/biogen-to-spend-2-5-billion-before-alzheimer-s-drug-results (accessed on 23 February 2021).
- Biogen. 221AD301 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT02477800 (accessed on 23 February 2021).
- A DiMasi, J.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, A.; Svensson, M.; Jacobi, F.; Allgulander, C.; Alonso, J.; Beghi, E.; Dodel, R.; Ekman, M.; Faravelli, C.; Fratiglioni, L.; et al. Cost of disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 718–779. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J. CNS Drugs Take Longer to Develop, Have Lower Success Rates, than Other Drugs. Tufts Center for the Study of Drug Development Website. 2014. Available online: http://csdd.tufts.edu/news/complete_story/pr_ir_nov_dec_ir (accessed on 23 February 2021).
- Gribkoff, V.K.; Kaczmarek, L.K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 2017, 120, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Take, C.D. 20% Longer to Develop and to Approve vs. Non-CNS Drugs. Tufts CSDD Impact Rep. 2018, 20, 5–8. [Google Scholar]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and Treatment Options. P T A Peer-Rev. J. Formul. Manag. 2014, 39, 638–645. [Google Scholar]
- Cunningham, C.; Wilcockson, D.C.; Boche, D.; Perry, V.H. Comparison of Inflammatory and Acute-Phase Responses in the Brain and Peripheral Organs of the ME7 Model of Prion Disease. J. Virol. 2005, 79, 5174–5184. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Nirmalraj, P.N.; List, J.; Battacharya, S.; Howe, G.; Xu, L.; Thompson, D.; Mayer, M. Complete aggregation pathway of amyloid β (1-40) and (1-42) resolved on an atomically clean interface. Sci. Adv. 2020, 6, eaaz6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Heegaard, N.H.H. Analysis of Protein Aggregation in Neurodegenerative Disease. Anal. Chem. 2013, 85, 4215–4227. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Morrison, B.M. Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Exp. Neurol. 2018, 309, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, T.N.; Athar, T.; Parveen, R.; Fatima, S. A review on protein misfolding, aggregation and strategies to prevent related ailments. Int. J. Biol. Macromol. 2017, 105, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, D.; de Deyn, P.P. Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef]
- Neha; Sodhi, R.K.; Jaggi, A.S.; Singh, N. Animal models of dementia and cognitive dysfunction. Life Sci. 2014, 109, 73–86. [Google Scholar] [CrossRef]
- Bracken, M.B. Why animal studies are often poor predictors of human reactions to exposure. J. R. Soc. Med. 2009, 102, 120–122. [Google Scholar] [CrossRef] [Green Version]
- Greek, R.; Menache, A. Systematic Reviews of Animal Models: Methodology versus Epistemology. Int. J. Med Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide—Identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ajay, G.W.B.; Murcko, M.A. Designing libraries with CNS activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Oldendorf, W.H.; Cancilla, P.; Frank, H.J.L. Blood-Brain Barrier: Interface between Internal Medicine and the Brain. Ann. Intern. Med. 1986, 105, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s Disease Facts and Figures. In Alzheimer’s Dementia; Wiley: New Jersey, NY, USA, 2020; pp. 391–460. [Google Scholar]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. In Alzheimer’s Dementia; Wiley: New Jersey, NY, USA, 2018. [Google Scholar]
- Rolfes, L.; Pawlitzki, M.; Pfeuffer, S.; Huntemann, N.; Wiendl, H.; Ruck, T.; Meuth, S.G. Failed, Interrupted, or Inconclusive Trials on Immunomodulatory Treatment Strategies in Multiple Sclerosis: Update 2015–2020. BioDrugs 2020, 34, 587–610. [Google Scholar] [CrossRef]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Kivipelto, M. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- Olanow, C.W.; Kieburtz, K.; Schapira, A.H. Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 64, S101–S110. [Google Scholar] [CrossRef]
- Da Costa, M.M.J.; Allen, C.E.; Higginbottom, A.; Ramesh, T.; Shaw, P.J.; McDermott, C.J. A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis. Model. Mech. 2013, 7, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGown, A.; McDearmid, J.R.; Panagiotaki, N.; Tong, H.; Al Mashhadi, S.; Redhead, N.; Lyon, A.N.; Beattie, C.E.; Shaw, P.J.; Ramesh, T.M. Early interneuron dysfunction in ALS: Insights from a mutant sod1 zebrafish model. Ann. Neurol. 2013, 73, 246–258. [Google Scholar] [CrossRef]
- Benedetti, L.; Ghilardi, A.; Rottoli, E.; De Maglie, M.; Prosperi, L.; Perego, C.; Francolini, M.I. NaP selective inhibition reverts precocious inter-and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model. Sci. Rep. 2016, 6, 1–20. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Zhang, P.-H.; Chen, Y.-J.; Wu, C.-L.; Tsai, H.-J. Conditional Overexpression of rtn4al in Muscle of Adult Zebrafish Displays Defects Similar to Human Amyotrophic Lateral Sclerosis. Mar. Biotechnol. 2018, 21, 52–64. [Google Scholar] [CrossRef]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Bugel, S.M.; Tanguay, R.L.; Planchart, A. Zebrafish: A Marvel of High-Throughput Biology for 21st Century Toxicology. Curr. Environ. Health Rep. 2014, 1, 341–352. [Google Scholar] [CrossRef] [Green Version]
- McGown, A.; Shaw, D.P.J.; Ramesh, T. ZNStress: A high-throughput drug screening protocol for identification of compounds modulating neuronal stress in the transgenic mutant sod1G93R zebrafish model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2016, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sager, J.J.; Bai, Q.; Burton, E.A. Transgenic zebrafish models of neurodegenerative diseases. Brain Struct. Funct. 2010, 214, 285–302. [Google Scholar] [CrossRef]
- De Abreu, M.S.; Friend, A.J.; Demin, K.A.; Amstislavskaya, T.G.; Bao, W.; Kalueff, A.V. Zebrafish models: Do we have valid paradigms for depression? J. Pharmacol. Toxicol. Methods 2018, 94, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, B. Sydney Brenner on the Genetics of Caenorhabditis elegans. Genetics 2016, 204, 1–2. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Carretero, M.; Solis, G.M.; Petrascheck, M.C. elegans as Model for Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Parker, J.A. Worming forward: Amyotrophic lateral sclerosis toxicity mechanisms and genetic interactions in Caenorhabditis elegans. Front. Genet. 2014, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.P. Dissecting Age Associated Disease in C. elegans. 2013. Available online: https://digital.wpi.edu/concern/student_works/4f16c444r?locale=en (accessed on 23 February 2021).
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s Disease in C. elegans. J. Parkinson’s Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [Green Version]
- Van Pelt, K.M.; Truttmann, M.C. Caenorhabditis elegans as a model system for studying aging-associated neurodegenerative diseases. Transl. Med. Aging 2020, 4, 60–72. [Google Scholar] [CrossRef]
- Solana-Manrique, C.; Moltó, M.D.; Calap-Quintana, P.; Sanz, F.J.; Llorens, J.V.; Paricio, N. Drosophila as a Model System for the Identification of Pharmacological Therapies in Neurodegenerative Diseases. In Insights into Human Neurodegeneration: Lessons Learnt from Drosophila; Springer International Publishing: New York, NY, USA, 2019; pp. 433–467. [Google Scholar]
- Sanz, F.J.; Solana-Manrique, C.; Muñoz-Soriano, V.; Calap-Quintana, P.; Moltó, M.D.; Paricio, N. Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic. Biol. Med. 2017, 108, 683–691. [Google Scholar] [CrossRef]
- Cho, Y.; Zhao, C.L.; Lu, H. Trends in high-throughput and functional neuroimaging inCaenorhabditis elegans. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1376. [Google Scholar] [CrossRef] [PubMed]
- Gosai, S.J.; Kwak, J.H.; Luke, C.J.; Long, O.S.; King, D.E.; Kovatch, K.J.; Johnston, P.A.; Shun, T.Y.; Lazo, J.S.; Perlmutter, D.H.; et al. Automated High-Content Live Animal Drug Screening Using C. elegans Expressing the Aggregation Prone Serpin α1-antitrypsin Z. PLoS ONE 2010, 5, e15460. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Hegarty, E.; Martin, C.; Gökçe, S.K.; Ghorashian, N.; Ben-Yakar, A. Large-scale microfluidics providing high-resolution and high-throughput screening of Caenorhabditis elegans poly-glutamine aggregation model. Nat. Commun. 2016, 7, 13023. [Google Scholar] [CrossRef]
- Johnson, T.E. Advantages and disadvantages of Caenorhabditis elegans for aging research. Exp. Gerontol. 2003, 38, 1329–1332. [Google Scholar] [CrossRef]
- Tissenbaum, H.A. Using C. elegans for aging research. Invertebr. Reprod. Develop. 2015, 59 (Suppl. S1), 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, C.D. Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol. Ther. 2006, 112, 677–700. [Google Scholar] [CrossRef]
- Nierode, G.J.; Perea, B.C.; McFarland, S.K.; Pascoal, J.F.; Clark, D.S.; Schaffer, D.V.; Dordick, J.S. High-Throughput Toxicity and Phenotypic Screening of 3D Human Neural Progenitor Cell Cultures on a Microarray Chip Platform. Stem Cell Rep. 2016, 7, 970–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-D.; Li, H.-Q.; Cui, M.; Dong, Q.; Yu, J.-T. Pluripotent stem cells for neurodegenerative disease modeling: An expert view on their value to drug discovery. Expert Opin. Drug Discov. 2020, 15, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Nilsson, O.; Strecker, R.; Lindvall, O.; Astedt, B.; Björklund, A. Behavioural effects of human fetal dopamine neurons grafted in a rat model of Parkinson’s disease. Exp. Brain Res. 1986, 65, 235–240. [Google Scholar] [CrossRef]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo-Arozena, A.; Kalmar, B.; Essa, S.; Ricketts, T.; Joyce, P.; Kent, R.; Rowe, C.; Parker, A.; Gray, A.; Hafezparast, M.; et al. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis. Model. Mech. 2011, 4, 686–700. [Google Scholar] [CrossRef] [Green Version]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Yang, W.; Yan, S.; Guo, X.; Li, X.-J. CRISPR/Cas9: A powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol. Neurodegener. 2015, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, K.; Kosic, M.; Lam, A.; Moradipour, A.; Zhao, Y.; Radisic, M. Biomaterials and Culture Systems for Development of Organoid and Organ-on-a-Chip Models. Ann. Biomed. Eng. 2020, 48, 2002–2027. [Google Scholar] [CrossRef]
- Lee, H.-K.; Sanchez, C.V.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; Van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood–brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.M.; Frega, M. Past, Present, and Future of Neuronal Models In Vitro. In Advances in Neurobiology; Springer International Publishing: New York, NY, USA, 2019; Volume 22, pp. 3–17. [Google Scholar]
- Gagliano, O.; Elvassore, N.; Luni, C. Microfluidic technology enhances the potential of human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 683–687. [Google Scholar] [CrossRef]
- Raasch, M.; Rennert, K.; Jahn, T.; Peters, S.; Henkel, T.; Huber, O.; Schulz, I.; Becker, H.; Lorkowski, S.; Funke, H.; et al. Microfluidically supported biochip design for culture of endothelial cell layers with improved perfusion conditions. Biofabrication 2015, 7, 015013. [Google Scholar] [CrossRef]
- Oddo, A.; Peng, B.; Tong, Z.; Wei, Y.; Tong, W.Y.; Thissen, H.; Voelcker, N.H. Advances in Microfluidic Blood–Brain Barrier (BBB) Models. Trends Biotechnol. 2019, 37, 1295–1314. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Bang, S.; Jeong, S.; Choi, N.; Kim, H.N. Brain-on-a-chip: A history of development and future perspective. Biomicrofluidics 2019, 13, 051301. [Google Scholar] [CrossRef]
- Chang, Y.; Kim, J.; Park, H.; Choi, H.; Kim, J. Modelling neurodegenerative diseases with 3D brain organoids. Biol. Rev. 2020, 95, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.H. Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab. Chip 2015, 15, 141–150. [Google Scholar] [CrossRef]
- Cho, H.; Hashimoto, T.; Wong, E.; Hori, Y.; Wood, L.B.; Zhao, L.; Haigis, K.M.; Hyman, B.T.; Irimia, D. Microfluidic Chemotaxis Platform for Differentiating the Roles of Soluble and Bound Amyloid-β on Microglial Accumulation. Sci. Rep. 2013, 3, srep01823. [Google Scholar] [CrossRef] [Green Version]
- Virlogeux, A.; Moutaux, E.; Christaller, W.; Genoux, A.; Bruyère, J.; Fino, E.; Charlot, B.; Cazorla, M.; Saudou, F. Reconstituting Corticostriatal Network on-a-Chip Reveals the Contribution of the Presynaptic Compartment to Huntington’s Disease. Cell Rep. 2018, 22, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Kaku, M. The Future of the Mind: The Scientific Quest to Understand, enhance, and Empower the Mind; Anchor Books; Doubleday: New York, NY, USA, 2015. [Google Scholar]
- Haston, K.M.; Finkbeiner, S. Clinical Trials in a Dish: The Potential of Pluripotent Stem Cells to Develop Therapies for Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 489–510. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Yuan, X.; Jones, Z.; Griffin, K.; Zhou, Y.; Ma, T.; Li, Y. Assembly of Human Stem Cell-Derived Cortical Spheroids and Vascular Spheroids to Model 3-D Brain-like Tissues. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating microglia from human pluripotent stem cells: Novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 2019, 14, 46–116. [Google Scholar] [CrossRef]
- Ormel, P.R.; De Sá, R.V.; Van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; Van Dijk, R.E.; Scheefhals, N.; Van Berlekom, A.B.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; A Banks, W.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- McManus, R.M.; Heneka, M.T. T cells in Alzheimer’s disease: Space invaders. Lancet Neurol. 2020, 19, 285–287. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Sette, A. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Town, T.; Tan, J.; Flavell, R.A.; Mullan, M. T-Cells in Alzheimer’s Disease. NeuroMolecular Med. 2005, 7, 255–264. [Google Scholar] [CrossRef]
- Heneka, M.T. An immune-cell signature marks the brain in Alzheimer’s disease. Nat. Cell Biol. 2020, 577, 322–323. [Google Scholar] [CrossRef] [Green Version]
- Pietronigro, E.; Zenaro, E.; Della Bianca, V.; Dusi, S.; Terrabuio, E.; Iannoto, G.; Slanzi, A.; Ghasemi, S.; Nagarajan, R.; Piacentino, G.; et al. Blockade of α4 integrins reduces leukocyte–endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Baik, S.H.; Cha, M.Y.; Hyun, Y.M.; Cho, H.; Hamza, B.; Kim, D.K.; Mook-Jung, I. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 2014, 35, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Mayorga, K.; Madariaga-Mazon, A.; Medina-Franco, J.L.; Maggiora, G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug Discov. 2020, 15, 293–306. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Gao, Y.; Zhu, G.; Wu, H.; Fang, Z.; Guo, K. Microfluidic synthesis of fatty acid esters: Integration of dynamic combinatorial chemistry and scale effect. Chem. Eng. J. 2020, 381, 122721. [Google Scholar] [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nat. Cell Biol. 2018, 557, S55–S57. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic–pharmacodynamic (PK–PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef]
- Michael, S.; Auld, D.; Klumpp, C.; Jadhav, A.; Zheng, W.; Thorne, N.; Austin, C.P.; Inglese, J.; Simeonov, A. A Robotic Platform for Quantitative High-Throughput Screening. ASSAY Drug Dev. Technol. 2008, 6, 637–657. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30. https://doi.org/10.3390/bioengineering8020030
Aldewachi H, Al-Zidan RN, Conner MT, Salman MM. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering. 2021; 8(2):30. https://doi.org/10.3390/bioengineering8020030
Chicago/Turabian StyleAldewachi, Hasan, Radhwan N. Al-Zidan, Matthew T. Conner, and Mootaz M. Salman. 2021. "High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases" Bioengineering 8, no. 2: 30. https://doi.org/10.3390/bioengineering8020030
APA StyleAldewachi, H., Al-Zidan, R. N., Conner, M. T., & Salman, M. M. (2021). High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering, 8(2), 30. https://doi.org/10.3390/bioengineering8020030