

Epigenetics and Gut Microbiota Crosstalk: A potential Factor in Pathogenesis of Cardiovascular Disorders

 ,
,  , and
, and 
Abstract
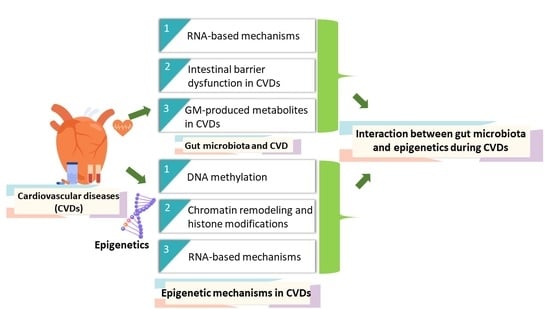
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Epigenetic Mechanisms in CVDs
2.1. DNA Methylation
2.2. Chromatin Remodelling and Histone Modifications
2.3. RNA-Based Mechanisms
3. Gut Microbiota and CVD: A Role of Heart-Gut Axis
3.1. RNA-Based Mechanisms
3.2. Intestinal Barrier Dysfunction in CVDs
3.3. GM-Produced Metabolites in CVDs
3.3.1. Bile Acids
3.3.2. Short-Chain Fatty Acids (SCFAs)
3.3.3. Trimethylamine N-Oxide (TMAO)
3.3.4. Phenylacetylglutamine (PAG)
4. Gut Microbiota and Epigenetics: Potential Interaction during CVDs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organizations. Cardiovascular Diseases (CVDs): Fact Sheet No 317. World Health Organizations. 2016. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 21 September 2022).
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Wilson, A.G. Epigenetic Regulation of Gene Expression in the Inflammatory Response and Relevance to Common Diseases. J. Periodontol. 2008, 79, 1514–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgeon, P.J.; Sukumar, A.N.; Marsden, P.A. Epigenetics of Cardiovascular Disease: A New ‘Beat’ in Coronary Artery Disease. Med Epigenet. 2014, 2, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Yang, S.; Lu, J.; Jin, X.; Wu, M. Diet-gut microbiota-epigenetics in metabolic diseases: From mechanisms to therapeutics. Biomed. Pharmacother. 2022, 153, 113290. [Google Scholar] [CrossRef]
- Paul, B.; Barnes, S.; Demark-Wahnefried, W.; Morrow, C.; Salvador, C.; Skibola, C.; Tollefsbol, T.O. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin. Epigenet. 2015, 7, 112. [Google Scholar] [CrossRef] [Green Version]
- Miro-Blanch, J.; Yanes, O. Epigenetic Regulation at the Interplay Between Gut Microbiota and Host Metabolism. Front. Genet. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Li, Y.; Stoll, M.L.; Tollefsbol, T.O. The Epigenetic Connection Between the Gut Microbiome in Obesity and Diabetes. Front. Genet. 2019, 10, 1329. [Google Scholar] [CrossRef]
- Shock, T.; Badang, L.; Ferguson, B.; Martinez-Guryn, K. The interplay between diet, gut microbes, and host epigenetics in health and disease. J. Nutr. Biochem. 2021, 95, 108631. [Google Scholar] [CrossRef] [PubMed]
- Duygu, B.; Poels, E.M.; da Costa Martins, P.A. Genetics and epigenetics of arrhythmia and heart failure. Front. Genet. 2013, 4, 219. [Google Scholar] [CrossRef] [Green Version]
- Wald, D.S.; Law, M.; Morris, J.K. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. BMJ 2002, 325, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiung, D.T.; Marsit, C.J.; Houseman, E.A.; Eddy, K.; Furniss, C.S.; McClean, M.D.; Kelsey, K.T. Global DNA Methylation Level in Whole Blood as a Biomarker in Head and Neck Squamous Cell Carcinoma. Cancer Epidemiol. Prev. Biomark. 2007, 16, 108–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foy, J.-P.; Pickering, C.R.; Papadimitrakopoulou, V.A.; Jelinek, J.; Lin, S.H.; William, W.N.; Frederick, M.J.; Wang, J.; Lang, W.; Feng, L. New DNA Methylation Markers and Global DNA Hypomethylation Are Associated with Oral Cancer Development. Cancer Prev. Res. 2015, 8, 1027–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, Q.; Zeng, F.; Li, W.; He, Z.; Chen, W.; Zhu, W.; Zhang, B. The Prognostic Value of Global DNA Hypomethylation in Cancer: A Meta-Analysis. PLoS ONE 2014, 9, e106290. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, S.; Su, Z.; Cheng, R.; Bai, X.; Li, X. LINE-1 Hypomethylation is Associated with the Risk of Coronary Heart Disease in Chinese Population. Arq. Bras. De Cardiol. 2014, 102, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-T.; Hsi, E.; Lin, H.-F.; Liao, Y.-C.; Wang, Y.-S.; Juo, S.-H.H. LINE-1 Methylation is Associated with an Increased Risk of Ischemic Stroke in Men. Curr. Neurovascular Res. 2014, 11, 4–9. [Google Scholar] [CrossRef]
- Baccarelli, A.; Wright, R.; Bollati, V.; Litonjua, A.; Zanobetti, A.; Tarantini, L.; Sparrow, D.; Vokonas, P.; Schwartz, J. Ischemic Heart Disease and Stroke in Relation to Blood DNA Methylation. Epidemiology (Camb. Mass.) 2010, 21, 819. [Google Scholar] [CrossRef]
- Guarrera, S.; Fiorito, G.; Onland-Moret, N.C.; Russo, A.; Agnoli, C.; Allione, A.; Di Gaetano, C.; Mattiello, A.; Ricceri, F.; Chiodini, P. Gene-specific DNA methylation profiles and LINE-1 hypomethylation are associated with myocardial infarction risk. Clin. Epigenet. 2015, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenvinkel, P.; Karimi, M.; Johansson, S.; Axelsson, J.; Suliman, M.; Lindholm, B.; Heimbürger, O.; Barany, P.; Alvestrand, A.; Nordfors, L. Impact of inflammation on epigenetic DNA methylation–A novel risk factor for cardiovascular disease? J. Intern. Med. 2007, 261, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Muka, T.; Koromani, F.; Portilla, E.; O’Connor, A.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Franco, O.H. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int. J. Cardiol. 2016, 212, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Mimi, C.Y.; Laird, P.W. DNA Methylation as a Biomarker for Cardiovascular Disease Risk. PLoS ONE 2010, 5, e9692. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Kumar, J.; Garg, G.; Kumar, A.; Patowary, A.; Karthikeyan, G.; Ramakrishnan, L.; Brahmachari, V.; Sengupta, S. Detection of Altered Global DNA Methylation in Coronary Artery Disease Patients. DNA Cell Biol. 2008, 27, 357–365. [Google Scholar] [CrossRef]
- Nguyen, A.; Mamarbachi, M.; Turcot, V.; Lessard, S.; Yu, C.; Luo, X.; Lalongé, J.; Hayami, D.; Gayda, M.; Juneau, M. Lower Methylation of the ANGPTL2 Gene in Leukocytes from Post-Acute Coronary Syndrome Patients. PLoS ONE 2016, 11, e0153920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Zhao, Y.; Zhang, Z.; Chen, J. Association of interleukin-6 methylation in leukocyte DNA with serum level and the risk of ischemic heart disease. Scand. J. Clin. Lab. Investig. 2016, 76, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Butts, B.; Gary, R.A.; Dunbar, S.B.; Butler, J. Methylation of Apoptosis-Associated Speck-Like Protein With a Caspase Recruitment Domain and Outcomes in Heart Failure. J. Card. Fail. 2016, 22, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Guay, S.-P.; Légaré, C.; Brisson, D.; Mathieu, P.; Bossé, Y.; Gaudet, D.; Bouchard, L. Epigenetic and genetic variations at the TNNT1 gene locus are associated with HDL-C levels and coronary artery disease. Epigenomics 2016, 8, 359–371. [Google Scholar] [CrossRef]
- Perkins, E.; Murphy, S.K.; Murtha, A.P.; Schildkraut, J.; Jirtle, R.L.; Demark-Wahnefried, W.; Forman, M.R.; Kurtzberg, J.; Overcash, F.; Huang, Z. Insulin-like growth factor 2/H19 methylation at birth and risk of overweight and obesity in children. J. Pediatr. 2012, 161, 31–39. [Google Scholar] [CrossRef]
- Deodati, A.; Inzaghi, E.; Liguori, A.; Puglianiello, A.; Germani, D.; Brufani, C.; Fintini, D.; Cappa, M.; Barbetti, F.; Cianfarani, S. IGF2 Methylation Is Associated with Lipid Profile in Obese Children. Horm. Res. Paediatr. 2013, 79, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Lee, S.; Lee, H.A.; Park, H.; Park, Y.J.; Ha, E.H.; Kim, Y.J. Can proopiomelanocortin methylation be used as an early predictor of metabolic syndrome? Diabetes Care 2014, 37, 734–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friso, S.; Pizzolo, F.; Choi, S.-W.; Guarini, P.; Castagna, A.; Ravagnani, V.; Carletto, A.; Pattini, P.; Corrocher, R.; Olivieri, O. Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis 2008, 199, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mou, S.; Pan, C. The FTO Gene rs9939609 Polymorphism Predicts Risk of Cardiovascular Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e71901. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Y.; Ma, J.; Guo, F.; Cao, Q.; Zhang, Y.; Zhou, B.; Chai, J.; Zhao, W.; Zhao, R. The Demethylase Activity of FTO (Fat Mass and Obesity Associated Protein) Is Required for Preadipocyte Differentiation. PLoS ONE 2015, 10, e0133788. [Google Scholar] [CrossRef] [Green Version]
- Rask-Andersen, M.; Almén, M.S.; Schiöth, H.B. Scrutinizing the FTO locus: Compelling evidence for a complex, long-range regulatory context. Hum. Genet. 2015, 134, 1183–1193. [Google Scholar] [CrossRef]
- Celis-Morales, C.; Marsaux, C.F.; Livingstone, K.M.; Navas-Carretero, S.; San-Cristobal, R.; O’Donovan, C.B.; Forster, H.; Woolhead, C.; Fallaize, R.; Macready, A.L. Physical activity attenuates the effect of the FTO genotype on obesity traits in European adults: The Food4Me study. Obesity 2016, 24, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Rönn, T.; Volkov, P.; Davegårdh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, A.; Nitert, M.D.; Eriksson, K.-F. A Six Months Exercise Intervention Influences the Genome-wide DNA Methylation Pattern in Human Adipose Tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef]
- Breitling, L.P.; Salzmann, K.; Rothenbacher, D.; Burwinkel, B.; Brenner, H. Smoking, F2RL3 methylation, and prognosis in stable coronary heart disease. Eur. Hear. J. 2012, 33, 2841–2848. [Google Scholar] [CrossRef]
- Talens, R.P.; Jukema, J.; Trompet, S.; Kremer, D.; Westendorp, R.; Lumey, L.; Sattar, N.; Putter, H.; Slagboom, P.; Heijmans, B. Hypermethylation at loci sensitive to the prenatal environment is associated with increased incidence of myocardial infarction. Int. J. Epidemiol. 2012, 41, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zheng, D.; Wang, L.; Huang, Y.; Liu, H.; Xu, L.; Liao, Q.; Liu, P.; Shi, X.; Wang, Z. Elevated PLA2G7 Gene Promoter Methylation as a Gender-Specific Marker of Aging Increases the Risk of Coronary Heart Disease in Females. PLoS ONE 2013, 8, e59752. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Peng, W.; Li, H.; Wang, W.; Wei, Y.; Li, W.; Xu, Y. Methylation of p15INK4b and Expression of ANRIL on Chromosome 9p21 Are Associated with Coronary Artery Disease. PLoS ONE 2012, 7, e47193. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Garg, G.; Kumar, A.; Mohammad, F.; Kumar, S.R.; Tanwar, V.S.; Sati, S.; Sharma, A.; Karthikeyan, G.; Brahmachari, V. Genome wide DNA methylation profiling for epigenetic alteration in coronary artery disease patients. Gene 2014, 541, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Guay, S.-P.; Brisson, D.; Mathieu, P.; Bossé, Y.; Gaudet, D.; Bouchard, L. A study in familial hypercholesterolemia suggests reduced methylomic plasticity in men with coronary artery disease. Epigenomics 2015, 7, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Úriz, A.M.; Milagro, F.I.; Mansego, M.L.; Cordero, P.; Abete, I.; De Arce, A.; Goyenechea, E.; Blázquez, V.; Martínez-Zabaleta, M.; Martínez, J.A. Obesity and ischemic stroke modulate the methylation levels of KCNQ1 in white blood cells. Hum. Mol. Genet. 2015, 24, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Movassagh, M.; Choy, M.-K.; Knowles, D.A.; Cordeddu, L.; Haider, S.; Down, T.; Siggens, L.; Vujic, A.; Simeoni, I.; Penkett, C. Distinct Epigenomic Features in End-Stage Failing Human Hearts. Circulation 2011, 124, 2411–2422. [Google Scholar] [CrossRef] [Green Version]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.M.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 413–429. [Google Scholar] [CrossRef]
- Zhang, Y.; Schöttker, B.; Florath, I.; Stock, C.; Butterbach, K.; Holleczek, B.; Mons, U.; Brenner, H. Smoking-Associated DNA Methylation Biomarkers and Their Predictive Value for All-Cause and Cardiovascular Mortality. Environ. Heal. Perspect. 2016, 124, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, S.; Cilluffo, R.; Prasad, M.; Best, P.J.; Atkinson, E.J.; Aoki, T.; Cunningham, J.M.; de Andrade, M.; Lerman, L.O.; Lerman, A. Sex-Specific Genetic Variants are Associated With Coronary Endothelial Dysfunction. J. Am. Heart Assoc. 2016, 5, e002544. [Google Scholar] [CrossRef]
- Zulfa, I.; Shim, E.B.; Song, K.-S.; Lim, K.M. Computational simulations of the effects of the G229D KCNQ1 mutation on human atrial fibrillation. J. Physiol. Sci. 2016, 66, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Irvin, M.R.; Zhi, D.; Joehanes, R.; Mendelson, M.; Aslibekyan, S.; Claas, S.A.; Thibeault, K.S.; Patel, N.; Day, K.; Jones, L.W. Epigenome-Wide Association Study of Fasting Blood Lipids in the Genetics of Lipid-Lowering Drugs and Diet Network Study. Circulation 2014, 130, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Sha, J.; Hidalgo, B.; Aslibekyan, S.; Do, A.N.; Zhi, D.; Sun, D.; Zhang, T.; Li, S.; Chen, W. Association of DNA Methylation at CPT1A Locus with Metabolic Syndrome in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) Study. PLoS ONE 2016, 11, e0145789. [Google Scholar] [CrossRef]
- Mamtani, M.; Kulkarni, H.; Dyer, T.D.; Göring, H.H.; Neary, J.L.; Cole, S.A.; Kent, J.W.; Kumar, S.; Glahn, D.C.; Mahaney, M.C. Genome- and epigenome-wide association study of hypertriglyceridemic waist in Mexican American families. Clin. Epigenetics 2016, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demerath, E.W.; Guan, W.; Grove, M.L.; Aslibekyan, S.; Mendelson, M.; Zhou, Y.-H.; Hedman, Å.K.; Sandling, J.K.; Li, L.-A.; Irvin, M.R. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum. Mol. Genet. 2015, 24, 4464–4479. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, F.; Aïssi, D.; Carrié, A.; Morange, P.-E.; Trégouët, D.-A. Robust validation of methylation levels association at CPT1A locus with lipid plasma levels. J. Lipid Res. 2014, 55, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriebel, J.; Herder, C.; Rathmann, W.; Wahl, S.; Kunze, S.; Molnos, S.; Volkova, N.; Schramm, K.; Carstensen-Kirberg, M.; Waldenberger, M. Association between DNA Methylation in Whole Blood and Measures of Glucose Metabolism: KORA F4 Study. PLoS ONE 2016, 11, e0152314. [Google Scholar] [CrossRef]
- Hidalgo, B.; Irvin, M.R.; Sha, J.; Zhi, D.; Aslibekyan, S.; Absher, D.; Tiwari, H.K.; Kabagambe, E.K.; Ordovas, J.M.; Arnett, D.K. Epigenome-Wide Association Study of Fasting Measures of Glucose, Insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network Study. Diabetes 2014, 63, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: A nested case-control study. Lancet Diabetes Endocrinol. 2015, 3, 526–534. [Google Scholar] [CrossRef]
- Pfeiffer, L.; Wahl, S.; Pilling, L.C.; Reischl, E.; Sandling, J.K.; Kunze, S.; Holdt, L.M.; Kretschmer, A.; Schramm, K.; Adamski, J. DNA Methylation of Lipid-Related Genes Affects Blood Lipid Levels. Circ. Cardiovasc. Genet. 2015, 8, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.M.; Wan, M.; Ding, J.; Taylor, J.R.; Lohman, K.; Su, D.; Bennett, B.D.; Porter, D.K.; Gimple, R.; Pittman, G.S. DNA Methylation of the Aryl Hydrocarbon Receptor Repressor Associations With Cigarette Smoking and Subclinical Atherosclerosis. Circ. Cardiovasc. Genet. 2015, 8, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Sayols, S.; Ganella, L.; Subirana Cachinero, I.; Salas, L.A.; Vilahur Chiaraviglio, N.; Corella, D.; Muñoz, D.; Segura, A.; Jiménez Conde, J.; Moran, S. Identification of a new locus and validation of previously reported loci showing differential methylation associated with smoking. The REGICOR study. Epigenetics 2015, 10, 1156–1165. [Google Scholar] [CrossRef]
- Koch, W.; Hoppmann, P.; De Waha, A.; Schömig, A.; Kastrati, A. Polymorphisms in thrombospondin genes and myocardial infarction: A case-control study and a meta-analysis of available evidence. Hum. Mol. Genet. 2008, 17, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Franklin, B.; Cascio, W.; Hong, Y.; Howard, G.; Lipsett, M.; Luepker, R.; Mittleman, M.; Samet, J.; Smith, S.C., Jr. Air pollution and cardiovascular disease: A statement for healthcare professionals from the expert panel on population and prevention science of the american heart association. Circulation 2004, 109, 2655–2671. [Google Scholar] [CrossRef]
- Panni, T.; Mehta, A.J.; Schwartz, J.D.; Baccarelli, A.A.; Just, A.C.; Wolf, K.; Wahl, S.; Cyrys, J.; Kunze, S.; Strauch, K. Genome-Wide Analysis of DNA Methylation and Fine Particulate Matter Air Pollution in Three Study Populations: KORA F3, KORA F4, and the Normative Aging Study. Environ. Health Perspect. 2016, 124, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Silva-Martínez, G.A.; Rodríguez-Ríos, D.; Alvarado-Caudillo, Y.; Vaquero, A.; Esteller, M.; Carmona, F.J.; Moran, S.; Nielsen, F.C.; Wickström-Lindholm, M.; Wrobel, K. Arachidonic and oleic acid exert distinct effects on the DNA methylome. Epigenetics 2016, 11, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, M.D.; Zeisel, S.H. Diet, methyl donors and DNA methylation: Interactions between dietary folate, methionine and choline. J. Nutr. 2002, 132, 2333S–2335S. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.-N.; Zhang, H.-P.; Sun, Y.; Yang, X.-L.; Wang, N.; Zhu, G.; Zhang, H.; Xu, H.; Ma, S.-C.; Zhang, Y. High-methionine diets accelerate atherosclerosis by HHcy-mediated FABP4 gene demethylation pathway via DNMT1 in ApoE−/− mice. FEBS Lett. 2015, 589, 3998–4009. [Google Scholar] [CrossRef] [Green Version]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef]
- Greißel, A.; Culmes, M.; Burgkart, R.; Zimmermann, A.; Eckstein, H.-H.; Zernecke, A.; Pelisek, J. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc. Pathol. 2015, 25, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Rong, S.; Repa, J.J.; Clair, R.S.; Parks, J.S.; Mishra, N. Histone Deacetylase 9 Represses Cholesterol Efflux and Alternatively Activated Macrophages in Atherosclerosis Development. Arter. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.T.; Araki, Y.; Sato, K.; Aizaki, Y.; Yokota, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem. Biophys. Res. Commun. 2014, 444, 682–686. [Google Scholar] [CrossRef]
- Gomez-Uriz, A.; Goyenechea, E.; Campion, J.; De Arce, A.; Martinez, M.; Puchau, B.; Milagro, F.; Abete, I.; Martínez, J.A.; Munain, A.L. Epigenetic patterns of two gene promoters (TNF-α and PON) in stroke considering obesity condition and dietary intake. J. Physiol. Biochem. 2014, 70, 603–614. [Google Scholar] [CrossRef]
- Lee, H.-A.; Lee, D.-Y.; Cho, H.-M.; Kim, S.-Y.; Iwasaki, Y.; Kim, I.K. Histone Deacetylase Inhibition Attenuates Transcriptional Activity of Mineralocorticoid Receptor Through Its Acetylation and Prevents Development of Hypertension. Circ. Res. 2013, 112, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Miao, X.; Liu, Y.; Li, F.; Liu, Q.; Sun, J.; Cai, L. Dysregulation of Histone Acetyltransferases and Deacetylases in Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2014, 2014, 641979. [Google Scholar] [CrossRef] [Green Version]
- Vadvalkar, S.S.; Baily, C.N.; Matsuzaki, S.; West, M.; Tesiram, Y.A.; Humphries, K.M. Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of Type 1 diabetes. Biochem. J. 2013, 449, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Tingare, A.; Thienpont, B.; Roderick, H.L. Epigenetics in the heart: The role of histone modifications in cardiac remodelling. Biochem. Soc. Trans. 2013, 41, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Thal, M.A.; Krishnamurthy, P.; Mackie, A.R.; Hoxha, E.; Lambers, E.; Verma, S.; Ramirez, V.; Qin, G.; Losordo, D.W.; Kishore, R. Enhanced Angiogenic and Cardiomyocyte Differentiation Capacity of Epigenetically Reprogrammed Mouse and Human Endothelial Progenitor Cells Augments Their Efficacy for Ischemic Myocardial Repair. Circ. Res. 2012, 111, 180–190. [Google Scholar] [CrossRef]
- Shen, J.; Han, X.; Ren, H.; Sun, W.; Gu, Y.; Qiao, J.; Dong, Q. Levels of histone H3 acetylation in peripheral blood mononuclear cells of acute cerebral infarction patients. Zhonghua Yi Xue Za Zhi 2014, 94, 2123–2128. [Google Scholar]
- Kaneda, R.; Takada, S.; Yamashita, Y.; Choi, Y.L.; Nonaka-Sarukawa, M.; Soda, M.; Misawa, Y.; Isomura, T.; Shimada, K.; Mano, H. Genome-wide histone methylation profile for heart failure. Genes Cells 2009, 14, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Tanios, F.; Reeps, C.; Zhang, J.; Schwamborn, K.; Eckstein, H.-H.; Zernecke, A.; Pelisek, J. Histone acetylation and histone acetyltransferases show significant alterations in human abdominal aortic aneurysm. Clin. Epigenet. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallaster, M.; Vallaster, C.D.; Wu, S.M. Epigenetic mechanisms in cardiac development and disease. Acta Biochim. Biophys. Sin. 2012, 44, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakova, Z.T.; Talaibekova, E.; Asambaeva, D.; Kerimkulova, A.; Lunegova, O.; Aldashev, A. Association of the polymorphic marker Glu23Lys in the KCNJ11 gene with hypertension in Kyrgyz patients. Ther. Arch. 2017, 89, 14–17. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Hobuß, L.; Bär, C.; Thum, T. Long non-coding RNAs: At the heart of cardiac dysfunction? Front. Physiol. 2019, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.-S.; Jin, J.-P.; Wang, J.-Q.; Zhang, Z.-G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Ahlin, F.; Arfvidsson, J.; Vargas, K.G.; Stojkovic, S.; Huber, K.; Wojta, J. MicroRNAs as circulating biomarkers in acute coronary syndromes: A review. Vasc. Pharmacol. 2016, 81, 15–21. [Google Scholar] [CrossRef]
- Bostjancic, E.; Zidar, N.; Stajner, D.; Glavac, D. MicroRNA miR-1 is up-regulated in remote myocardium in patients with myocardial infarction. Folia Biol. (Praha) 2010, 56, 27–31. [Google Scholar]
- Zhu, W.S.; Guo, W.; Zhu, J.N.; Tang, C.M.; Fu, Y.H.; Lin, Q.X.; Tan, N.; Shan, Z.X. Hsp90aa1: A novel target gene of miR-1 in cardiac ischemia/reperfusion injury. Sci. Rep. 2016, 6, 24498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Gui, Y.; Guo, Y.; Xu, D. The regulatory function of microRNA-1 in arrhythmias. Mol. BioSyst. 2016, 12, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M. The Role of MicroRNA-133 in Cardiac Hypertrophy Uncovered. Circ. Res. 2010, 106, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Antonicelli, R.; Lorenzi, M.; D’Alessandra, Y.; Lazzarini, R.; Santini, G.; Spazzafumo, L.; Lisa, R.; La Sala, L.; Galeazzi, R. Diagnostic potential of circulating miR-499-5p in elderly patients with acute non ST-elevation myocardial infarction. Int. J. Cardiol. 2013, 167, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Quiat, D.; Olson, E.N. MicroRNAs in cardiovascular disease: From pathogenesis to prevention and treatment. J. Clin. Investig. 2013, 123, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Takahashi, R.; Hiura, Y.; Hirokawa, G.; Fukushima, Y.; Iwai, N. Plasma miR-208 as a Biomarker of Myocardial Injury. Clin. Chem. 2009, 55, 1944–1949. [Google Scholar] [CrossRef] [Green Version]
- Di, Y.; Zhang, D.; Hu, T.; Li, D. miR-23 regulate the pathogenesis of patients with coronary artery disease. Int. J. Clin. Exp. Med. 2015, 8, 11759. [Google Scholar]
- Jin, Y.; Yang, C.-J.; Xu, X.; Cao, J.-N.; Feng, Q.-T.; Yang, J. MiR-214 regulates the pathogenesis of patients with coronary artery disease by targeting VEGF. Mol. Cell. Biochem. 2015, 402, 111–122. [Google Scholar] [CrossRef]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A. Cardiac fibroblast–derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef] [Green Version]
- Zampetaki, A.; Willeit, P.; Tilling, L.; Drozdov, I.; Prokopi, M.; Renard, J.-M.; Mayr, A.; Weger, S.; Schett, G.; Shah, A.; et al. Prospective Study on Circulating MicroRNAs and Risk of Myocardial Infarction. J. Am. Coll. Cardiol. 2012, 60, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Rotllan, N.; Canfrán-Duque, A.; Aranda, J.F.; Ramírez, C.M.; Araldi, E.; Lin, C.-S.; Anderson, N.N.; Wagschal, A.; de Cabo, R. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagschal, A.; Najafi-Shoushtari, S.H.; Wang, L.; Goedeke, L.; Sinha, S.; Delemos, A.S.; Black, J.C.; Ramírez, C.M.; Li, Y.; Tewhey, R. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015, 21, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Matute, J.; Rotllan, N.; Aranda, J.F.; Fernández-Hernando, C. MicroRNAs and atherosclerosis. Curr. Atheroscler. Rep. 2013, 15, 322. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Wu, C.; Khan, A.M.; Bloch, D.B.; Davis-Dusenbery, B.N.; Ghorbani, A.; Spagnolli, E.; Martinez, A.; Ryan, A.; Tainsh, L.T. Atrial natriuretic peptide is negatively regulated by microRNA-425. J. Clin. Investig. 2013, 123, 3378–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The Gut Microbiota as an Environmental Factor That Regulates Fat Storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Scademy Sci. 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027. [Google Scholar] [CrossRef]
- Cho, I.; Yamanishi, S.; Cox, L.; Methé, B.A.; Zavadil, J.; Li, K.; Gao, Z.; Mahana, D.; Raju, K.; Teitler, I. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 2012, 488, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 4592–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Skye, S.M.; Zhu, W.; Romano, K.A.; Guo, C.-J.; Wang, Z.; Jia, X.; Kirsop, J.; Haag, B.; Lang, J.M.; DiDonato, J.A. Microbial Transplantation With Human Gut Commensals Containing CutC Is Sufficient to Transmit Enhanced Platelet Reactivity and Thrombosis Potential. Circ. Res. 2018, 123, 1164–1176. [Google Scholar] [CrossRef]
- Ott, S.J.; El Mokhtari, N.E.; Musfeldt, M.; Hellmig, S.; Freitag, S.; Rehman, A.; Kuhbacher, T.; Nikolaus, S.; Namsolleck, P.; Blaut, M.; et al. Detection of Diverse Bacterial Signatures in Atherosclerotic Lesions of Patients With Coronary Heart Disease. Circulation 2006, 113, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Fåk, F.; Tremaroli, V.; Bergström, G.; Bäckhed, F. Oral Microbiota in Patients with Atherosclerosis. Atherosclerosis 2015, 243, 573–578. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziganshina, E.E.; Sharifullina, D.M.; Lozhkin, A.P.; Khayrullin, R.N.; Ignatyev, I.M.; Ziganshin, A.M. Bacterial Communities Associated with Atherosclerotic Plaques from Russian Individuals with Atherosclerosis. PLoS ONE 2016, 11, e0164836. [Google Scholar] [CrossRef] [PubMed]
- Lanter, B.B.; Sauer, K.; Davies, D.G. Bacteria Present in Carotid Arterial Plaques Are Found as Biofilm Deposits Which May Contribute to Enhanced Risk of Plaque Rupture. Mbio 2014, 5, e01206–e01214. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Brar, M.S.; Kirjavainen, P.V.; Chen, Y.; Peng, J.; Li, D.; Leung, F.C.; El-Nezami, H. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: A pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE(-/-) mice. BMC Microbiol. 2016, 16, 264. [Google Scholar]
- Stepankova, R.; Tonar, Z.; Bartova, J.; Nedorost, L.; Rossman, P.; Poledne, R.; Schwarzer, M.; Tlaskalova-Hogenova, H. Absence of Microbiota (Germ-Free Conditions) Accelerates the Atherosclerosis in ApoE-Deficient Mice Fed Standard Low Cholesterol Diet. J. Atheroscler. Thromb. 2010, 17, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Tanoue, T.; Yamashita, T.; Yodoi, K.; Matsumoto, T.; Emoto, T.; Mizoguchi, T.; Hayashi, T.; Kitano, N.; Sasaki, N.; et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J. Lipid Res. 2017, 58, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Kramer, C.D.; Simas, A.M.; He, X.; Ingalls, R.R.; Weinberg, E.O.; Genco, C.A. Distinct roles for dietary lipids and Porphyromonas gingivalis infection on atherosclerosis progression and the gut microbiota. Anaerobe 2017, 45, 19–30. [Google Scholar] [CrossRef]
- Calandrini, C.A.; Ribeiro, A.C.; Gonnelli, A.C.; Ota-Tsuzuki, C.; Rangel, L.P.; Saba-Chujfi, E.; Mayer, M.P. Microbial composition of atherosclerotic plaques. Oral Dis. 2014, 20, e128–e134. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.A.; Rosenfeld, M. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef]
- Polsinelli, V.B.; Sinha, A.; Shah, S.J. Visceral Congestion in Heart Failure: Right Ventricular Dysfunction, Splanchnic Hemodynamics, and the Intestinal Microenvironment. Curr. Hear. Fail. Rep. 2017, 14, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Munger, M.A.; Johnson, B.; Amber, I.J.; Callahan, K.S.; Gilbert, E.M. Circulating concentrations of proinflammatory cytolcines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1996, 77, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Rauchhaus, M.; Doehner, W.; Francis, D.P.; Davos, C.; Kemp, M.; Liebenthal, C.; Niebauer, J.; Hooper, J.; Volk, H.-D.; Coats, A.J. Plasma Cytokine Parameters and Mortality in Patients With Chronic Heart Failure. Circulation 2000, 102, 3060–3067. [Google Scholar] [CrossRef] [PubMed]
- Hug, H.; Mohajeri, M.H.; La Fata, G. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients 2018, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Niebauer, J.; Volk, H.D.; Kemp, M.; Dominguez, M.; Schumann, R.R.; Rauchhaus, M.; Poole-Wilson, P.A.; Coats, A.J.; Anker, S.D. Endotoxin and immune activation in chronic heart failure: A prospective cohort study. Lancet 1999, 353, 1838–1842. [Google Scholar] [CrossRef]
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-Derived Serum Lipopolysaccharide is Associated With Enhanced Risk of Major Adverse Cardiovascular Events in Atrial Fibrillation: Effect of Adherence to Mediterranean Diet. J. Am. Hear. Assoc. 2017, 6, e005784. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Clements, W.; Parks, R.; Erwin, P.; Halliday, M.; Barr, J.; Rowlands, B. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut 1996, 39, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; Astiarraga, B.; Camastra, S.; Accili, D.; Ferrannini, E. Human Insulin Resistance Is Associated With Increased Plasma Levels of 12α-Hydroxylated Bile Acids. Diabetes 2013, 62, 4184–4191. [Google Scholar] [CrossRef] [Green Version]
- Choucair, I.; Nemet, I.; Li, L.; Cole, M.A.; Skye, S.M.; Kirsop, J.D.; Fischbach, M.A.; Gogonea, V.; Brown, J.M.; Tang, W.W.; et al. Quantification of bile acids: A mass spectrometry platform for studying gut microbe connection to metabolic diseases. J. Lipid Res. 2020, 61, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wang, X.; Li, J.; Zhang, Y.; Zhong, H.; Liu, R.; Zhang, D.; Feng, Q.; Xie, X.; Hong, J. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat. Commun. 2017, 8, 1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deehan, E.C.; Yang, C.; Perez-Muñoz, M.E.; Nguyen, N.K.; Cheng, C.C.; Triador, L.; Zhang, Z.; Bakal, J.A.; Walter, J. Precision Microbiome Modulation with Discrete Dietary Fiber Structures Directs Short-Chain Fatty Acid Production. Cell Host Microbe 2020, 27, 389–404. [Google Scholar] [CrossRef]
- Miller, T.L.; Wolin, M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Ramesh, V.; Locasale, J.W. Acetate Metabolism in Physiology, Cancer, and Beyond. Trends Cell Biol. 2019, 29, 695–703. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef] [Green Version]
- Nobel, Y.R.; Cox, L.M.; Kirigin, F.F.; Bokulich, N.A.; Yamanishi, S.; Teitler, I.; Chung, J.; Sohn, J.; Barber, C.M.; Goldfarb, D.S. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 2015, 6, 7486. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, V.E.; Battaglia, T.; Kurtz, Z.D.; Bijnens, L.; Ou, A.; Engstrand, I.; Zheng, X.; Iizumi, T.; Mullins, B.J.; Müller, C.L. A single early-in-life macrolide course has lasting effects on murine microbial network topology and immunity. Nat. Commun. 2017, 8, 518. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-S.; Li, J.; Krautkramer, K.A.; Badri, M.; Battaglia, T.; Borbet, T.; Koh, H.; Ng, S.; Sibley, R.A.; Li, Y. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. Elife 2018, 7, e37816. [Google Scholar] [CrossRef] [PubMed]
- Livanos, A.E.; Greiner, T.U.; Vangay, P.; Pathmasiri, W.; Stewart, D.; McRitchie, S.; Li, H.; Chung, J.; Sohn, J.; Kim, S. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat. Microbiol. 2016, 1, 16140. [Google Scholar] [CrossRef] [PubMed]
- Whelton, S.P.; Hyre, A.D.; Pedersen, B.; Yi, Y.; Whelton, P.K.; He, J. Effect of dietary fiber intake on blood pressure: A meta-analysis of randomized, controlled clinical trials. J. Hypertens. 2005, 23, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T. Olfactory receptor responding to gut microbiota derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Poll, B.; Steppan, J.; Lester, L.; Berkowitz, D.; Pluznick, J. A short chain fatty acid produced by the gut microbiota plays a role in blood pressure regulation and cardiac contractility. FASEB J. 2019, 33, 569.19-569.19. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Mell, B.; Jala, V.R.; Mathew, A.V.; Byun, J.; Waghulde, H.; Zhang, Y.; Haribabu, B.; Vijay-Kumar, M.; Pennathur, S.; Joe, B. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol. Genom. 2015, 47, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Markó, L.; Höges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G. Short-Chain Fatty Acid Propionate Protects From Hypertensive Cardiovascular Damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.W.; Chen, H.-C.; Chen, C.-Y.; Yen, C.Y.; Lin, C.-J.; Prajnamitra, R.P.; Chen, L.-L.; Ruan, S.-C.; Lin, J.-H.; Lin, P.-J. Loss of Gut Microbiota Alters Immune System Composition and Cripples Postinfarction Cardiac Repair. Circulation 2019, 139, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Battson, M.L.; Lee, D.M.; Li Puma, L.C.; Ecton, K.E.; Thomas, K.N.; Febvre, H.P.; Chicco, A.J.; Weir, T.L.; Gentile, C.L. Gut microbiota regulates cardiac ischemic tolerance and aortic stiffness in obesity. Am. J. Physiol. Circ. Physiol. 2019, 317, H1210–H1220. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D. γ-Butyrobetaine Is a Proatherogenic Intermediate in Gut Microbial Metabolism of L-Carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.S.; Wang, Z.; Cajka, T.; Buffa, J.A.; Nemet, I.; Hurd, A.G.; Gu, X.; Skye, S.M.; Roberts, A.B.; Wu, Y.; et al. Untargeted metabolomics identifies trimethyllysine, a TMAO-producing nutrient precursor, as a predictor of incident cardiovascular disease risk. J. Clin. Investig. 2018, 3, e99096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Aldana-Hernández, P.; Leonard, K.-A.; Zhao, Y.-Y.; Curtis, J.M.; Field, C.J.; Jacobs, R.L. Dietary choline or trimethylamine N-oxide supplementation does not influence atherosclerosis development in LDLR−/− and ApoE−/− male mice. J. Nutr. 2020, 150, 249–255. [Google Scholar] [CrossRef]
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018, 17, 286. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Chen, J.; Chen, J.; Tao, J.; Wu, S.; Xu, G.; Wang, Z.; Wei, D.; Yin, W. Trimethylamine N-oxide promotes apoE −/− mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J. Cell. Physiol. 2020, 235, 6582–6591. [Google Scholar] [CrossRef]
- Xue, J.; Zhou, D.; Poulsen, O.; Imamura, T.; Hsiao, Y.-H.; Smith, T.H.; Malhotra, A.; Dorrestein, P.; Knight, R.; Haddad, G.G. Intermittent Hypoxia and Hypercapnia Accelerate Atherosclerosis, Partially via Trimethylamine-Oxide. Am. J. Respir. Cell Mol. Biol. 2017, 57, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe–targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Zhu, W.; Buffa, J.; Wang, Z.; Warrier, M.; Schugar, R.; Shih, D.; Gupta, N.; Gregory, J.; Org, E.; Fu, X. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J. Thromb. Haemost. 2018, 16, 1857–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-κB (Nuclear Factor κB) Signals. Arter. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef]
- Jin, B.; Ji, F.; Zuo, A.; Liu, H.; Qi, L.; He, Y.; Wang, Q.; Zhao, P. Destructive Role of TMAO in T-Tubule and Excitation-Contraction Coupling in the Adult Cardiomyocytes. Int. Hear. J. 2020, 61, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Organ, C.L.; Otsuka, H.; Bhushan, S.; Wang, Z.; Bradley, J.; Trivedi, R.; Polhemus, D.J.; Tang, W.W.; Wu, Y.; Hazen, S.L. Choline diet and its gut microbe–derived metabolite, trimethylamine N-oxide, exacerbate pressure overload–induced heart failure. Circ. Heart Fail. 2016, 9, e002314. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Kong, B.; Shuai, W.; Fu, H.; Jiang, X.; Huang, H. 3,3-Dimethyl-1-butanol attenuates cardiac remodeling in pressure-overload-induced heart failure mice. J. Nutr. Biochem. 2020, 78, 108341. [Google Scholar] [CrossRef]
- Gupta, N.; Buffa, J.A.; Roberts, A.B.; Sangwan, N.; Skye, S.M.; Li, L.; Ho, K.J.; Varga, J.; DiDonato, J.A.; Tang, W.W. Targeted Inhibition of Gut Microbial Trimethylamine N-Oxide Production Reduces Renal Tubulointerstitial Fibrosis and Functional Impairment in a Murine Model of Chronic Kidney Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 1239–1255. [Google Scholar] [CrossRef] [PubMed]
- Nanto-Hara, F.; Kanemitsu, Y.; Fukuda, S.; Kikuchi, K.; Asaji, K.; Saigusa, D.; Iwasaki, T.; Ho, H.-J.; Mishima, E.; Suzuki, T. The guanylate cyclase C agonist linaclotide ameliorates the gut–cardio–renal axis in an adenine-induced mouse model of chronic kidney disease. Nephrol. Dial. Transplant. 2020, 35, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Wang, Z.; Fan, Y.; Wu, Y.; Hazen, S.L.; Tang, W.W. Trimethylamine N -Oxide and Mortality Risk in Patients With Peripheral Artery Disease. J. Am. Hear. Assoc. 2016, 5, e004237. [Google Scholar] [CrossRef]
- Senthong, V.; Li, X.S.; Hudec, T.; Coughlin, J.; Wu, Y.; Levison, B.; Wang, Z.; Hazen, S.L.; Tang, W.W. Plasma trimethylamine N-oxide, a gut microbe–generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J. Am. Coll. Cardiol. 2016, 67, 2620–2628. [Google Scholar] [CrossRef]
- Tan, Y.; Sheng, Z.; Zhou, P.; Liu, C.; Zhao, H.; Song, L.; Li, J.; Zhou, J.; Chen, Y.; Wang, L. Plasma Trimethylamine N-Oxide as a Novel Biomarker for Plaque Rupture in Patients With ST-Segment–Elevation Myocardial Infarction. Circ. Cardiovasc. Interv. 2019, 12, e007281. [Google Scholar] [CrossRef]
- Lever, M.; George, P.M.; Slow, S.; Bellamy, D.; Young, J.M.; Ho, M.; McEntyre, C.J.; Elmslie, J.L.; Atkinson, W.; Molyneux, S.L. Betaine and Trimethylamine-N-Oxide as Predictors of Cardiovascular Outcomes Show Different Patterns in Diabetes Mellitus: An Observational Study. PLoS ONE 2014, 9, e114969. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Heaney, L.M.; Bhandari, S.S.; Jones, D.J.L.; Ng, L.L. TrimethylamineN-oxide and prognosis in acute heart failure. Heart 2016, 102, 841–848. [Google Scholar] [CrossRef] [Green Version]
- Trøseid, M.; Ueland, T.; Hov, J.; Svardal, A.; Gregersen, I.; Dahl, C.; Aakhus, S.; Gude, E.; Bjørndal, B.; Halvorsen, B. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e822. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of Platelet Function Through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gareri, C.; Rockman, H.A. G-protein–coupled receptors in heart disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Honour, J. The possible involvement of intestinal bacteria in steroidal hypertension. Endocrinology 1982, 110, 285–287. [Google Scholar] [CrossRef]
- Jäckel, S.; Kiouptsi, K.; Lillich, M.; Hendrikx, T.; Khandagale, A.; Kollar, B.; Hörmann, N.; Reiss, C.; Subramaniam, S.; Wilms, E. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood J. Am. Soc. Hematol. 2017, 130, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Padanilam, B.J. Renal Nerves Drive Interstitial Fibrogenesis in Obstructive Nephropathy. J. Am. Soc. Nephrol. 2012, 24, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Lund, R.; Laiho, A.; Lundelin, K.; Ley, R.E.; Isolauri, E.; Salminen, S. Gut Microbiota as an Epigenetic Regulator: Pilot Study Based on Whole-Genome Methylation Analysis. mBio 2014, 5, e02113-14. [Google Scholar] [CrossRef] [Green Version]
- Shenderov, B.A. Gut indigenous microbiota and epigenetics. Microb. Ecol. Health Dis. 2012, 23, 17195. [Google Scholar] [CrossRef]
- Stilling, R.M.; Dinan, T.G.; Cryan, J.F. Microbial genes, brain & behaviour–epigenetic regulation of the gut–brain axis. Genes Brain Behav. 2014, 13, 69–86. [Google Scholar]
- Watson, M.M.; Søreide, K. The gut microbiota influence on human epigenetics, health, and disease. In Handbook of Epigenetics: The New Molecular and Medical Genetics; Elsevier Academic Press: Cambridge, MA, USA, 2017; pp. 495–510. [Google Scholar]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut Microbiota Composition and Activity in Relation to Host Metabolic Phenotype and Disease Risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Battson, M.L.; Lee, D.M.; Weir, T.L.; Gentile, C.L. The gut microbiota as a novel regulator of cardiovascular function and disease. J. Nutr. Biochem. 2018, 56, 1–15. [Google Scholar] [CrossRef]
- Abrahamsson, T.R.; Wu, R.Y.; Jenmalm, M.C. Gut microbiota and allergy: The importance of the pregnancy period. Pediatr. Res. 2014, 77, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, J. The microbiome during pregnancy and early postnatal life. Semin. Fetal Neonatal Med. 2016, 21, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A. Microbiota of the Human Body: Implications in Health and Disease; Springer International Publishing AG: Cham, Switzerland, 2016; pp. 154–196. [Google Scholar]
- Bhat, M.I.; Kapila, R. Dietary metabolites derived from gut microbiota: Critical modulators of epigenetic changes in mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary Gut Microbial Metabolites, Short-chain Fatty Acids, and Host Metabolic Regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef]
- Li, G.; Yao, W.; Jiang, H. Short-Chain Fatty Acids Enhance Adipocyte Differentiation in the Stromal Vascular Fraction of Porcine Adipose Tissue. J. Nutr. 2014, 144, 1887–1895. [Google Scholar] [CrossRef] [Green Version]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential Modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of Host Peripheral Lipid Metabolism and Histone Acetylation in Mouse Gut Organoids. Mbio 2014, 5, e01438-14. [Google Scholar] [CrossRef] [Green Version]
- Remely, M.; Lovrecic, L.; De La Garza, A.; Migliore, L.; Peterlin, B.; Milagro, F.; Martinez, A.; Haslberger, A. Therapeutic perspectives of epigenetically active nutrients. Br. J. Pharmacol. 2015, 172, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Burns, M.B.; Subramanian, S.; Blekhman, R. Interaction between Host MicroRNAs and the Gut Microbiota in Colorectal Cancer. mSystems® 2018, 3, e00205-17. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, V.; Nagu, P.; Inbaraj, B.S.; Sharma, M.; Parashar, A.; Sridhar, K. Epigenetics and Gut Microbiota Crosstalk: A potential Factor in Pathogenesis of Cardiovascular Disorders. Bioengineering 2022, 9, 798. https://doi.org/10.3390/bioengineering9120798
Mehta V, Nagu P, Inbaraj BS, Sharma M, Parashar A, Sridhar K. Epigenetics and Gut Microbiota Crosstalk: A potential Factor in Pathogenesis of Cardiovascular Disorders. Bioengineering. 2022; 9(12):798. https://doi.org/10.3390/bioengineering9120798
Chicago/Turabian StyleMehta, Vineet, Priyanka Nagu, Baskaran Stephen Inbaraj, Minaxi Sharma, Arun Parashar, and Kandi Sridhar. 2022. "Epigenetics and Gut Microbiota Crosstalk: A potential Factor in Pathogenesis of Cardiovascular Disorders" Bioengineering 9, no. 12: 798. https://doi.org/10.3390/bioengineering9120798
APA StyleMehta, V., Nagu, P., Inbaraj, B. S., Sharma, M., Parashar, A., & Sridhar, K. (2022). Epigenetics and Gut Microbiota Crosstalk: A potential Factor in Pathogenesis of Cardiovascular Disorders. Bioengineering, 9(12), 798. https://doi.org/10.3390/bioengineering9120798