
Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea


{kind=link}
Abstract
:1. Summary
2. Data Description
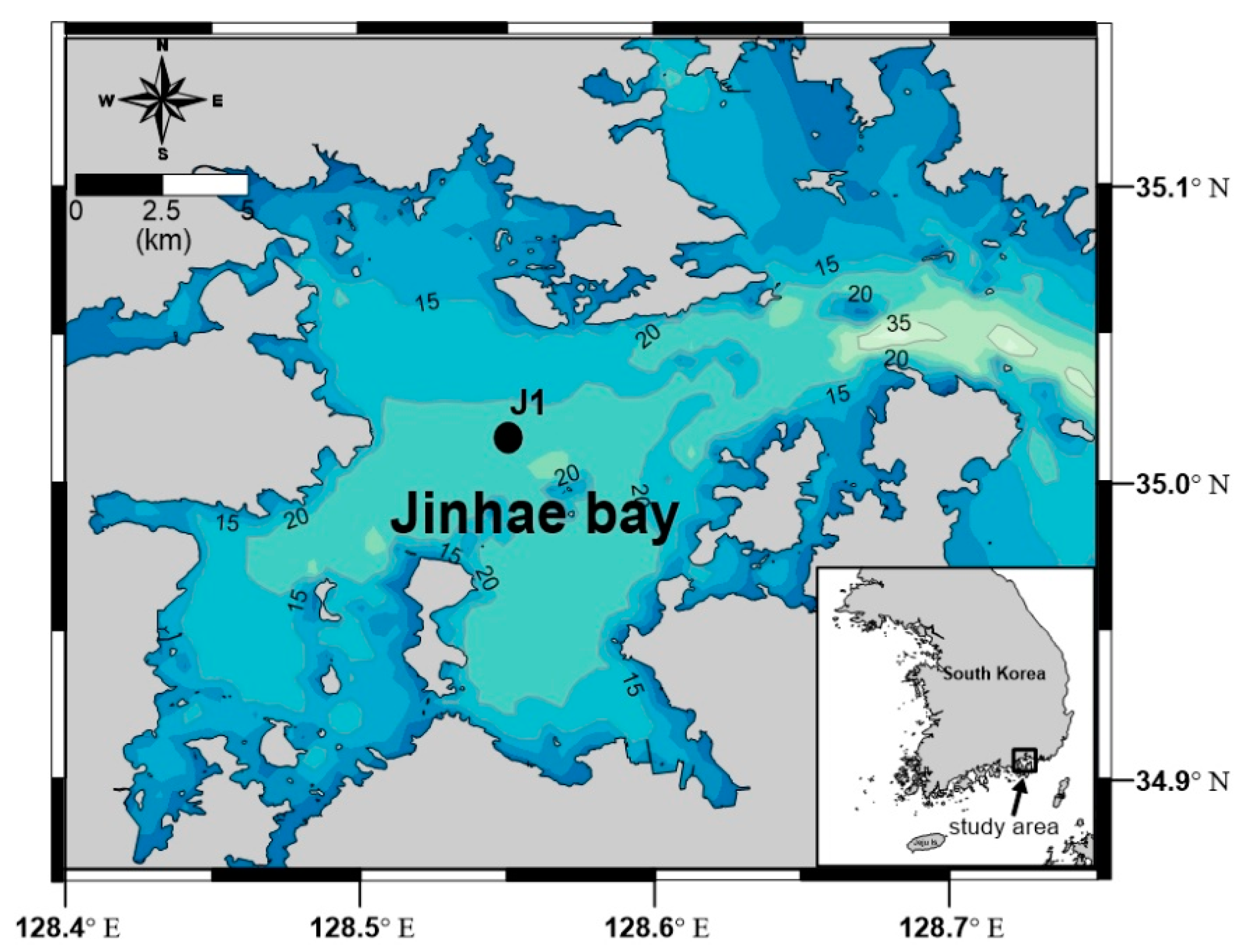
2.1. Study Area
2.2. Bacterial Community Compositions
3. Methods
3.1. Sample Collection
3.2. Measurement of Marine Environmental Variables
3.3. DNA Extraction and Sequencing
3.4. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, D.; Choi, H.-W.; Choi, S.-H.; Baek, S.H.; Kim, K.-H.; Jeong, J.-H.; Kim, Y. Spatial and seasonal variations in the water quality of Jinhae Bay, Korea. N. Z. J. Mar. Freshwater Res. 2013, 47, 192–207. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-S.; Lee, Y.-H.; Kwon, J.-N.; Choi, H.-G. The effect of low oxygen conditions on biogeo- chemical cycling of nutrients in a shallow seasonally stratified bay in southeast Korea (Jinhae Bay). Mar. Pollut. Bull. 2015, 95, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-K.; Park, T.-G.; Park, Y.-T.; Lim, W.-A. Monitoring and trends in harmful algal blooms and red tides in Korean coastal waters, with emphasis on Cochlodinium polykrikoides. Harmful Algae 2013, 30, S3–S14. [Google Scholar] [CrossRef]
- Lim, H.-S.; Diaz, R.J.; Hong, J.-S.; Schaffner, L.C. Hypoxia and benthic community recovery in Korean coastal waters. Mar. Pollut. Bull. 2006, 52, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Lee, S.H.; Park, J.; Lee, J.; Yoon, J.-E.; Kim, I.-N. Coastal hypoxia in the Jinhae bay, South Korea: Mechanism, spatiotemporal variation, and implications (based on 2011 survey). J. Coast. Res. 2018, 85, 1481–1485. [Google Scholar] [CrossRef]
- Cho, C.H. Mass mortalities of oyster due to red tide in Jinhae Bay in 1978. Korean J. Fish. Aquat. Sci. 1979, 12, 27–33. [Google Scholar]
- Lee, J.; Park, K.-T.; Lim, J.-H.; Yoon, J.-E.; Kim, I.-N. Hypoxia in Korean coastal waters: A case study of the natural Jinhae Bay and artificial Shihwa Bay. Front. Mar. Sci. 2018, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids. Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Chapter Nineteen—Advancing Our Understanding of the Human Microbiome Using QIIME. In Methods in Enzymology; DeLong, E.F., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 531, pp. 371–444. [Google Scholar]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 10, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, E0227434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yu, W.-H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010, 2010, baq013. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2009, 26, 266–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soergel, D.A.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.-H.; Kim, I.-N. Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea. Data 2021, 6, 44. https://doi.org/10.3390/data6050044
Lim J-H, Kim I-N. Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea. Data. 2021; 6(5):44. https://doi.org/10.3390/data6050044
Chicago/Turabian StyleLim, Jae-Hyun, and Il-Nam Kim. 2021. "Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea" Data 6, no. 5: 44. https://doi.org/10.3390/data6050044
APA StyleLim, J. -H., & Kim, I. -N. (2021). Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea. Data, 6(5), 44. https://doi.org/10.3390/data6050044