

H3K36 Di-Methylation Marks, Mediated by Ash1 in Complex with Caf1-55 and MRG15, Are Required during Drosophila Heart Development

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drosophila Lines
2.2. Lethality at Eclosion
2.3. Single-Cell RNA Sequencing: Embryo Collection and Cell Isolation
2.4. Single-Cell RNA Sequencing: Library Generation and Sequencing
2.5. Single-Cell RNA Sequencing: Data Processing and Cardiogenic Progenitors Cluster Analysis
2.6. Confirmation RNAi by Quantitative RT-PCR
2.7. Adult Drosophila Survival Assay
2.8. Heart Structural Analysis: Immunochemistry
2.9. Heart Structural Analysis: Quantitation
2.10. OCT Measurements and Analysis of Cardiac Function
2.11. H3K36 Methylation Marks: Corroborate Antibody Detection
2.12. H3K36 Methylation Marks: Immunochemistry
2.13. H3K36 Methylation Marks: Quantitation
2.14. Statistical Analysis
3. Results
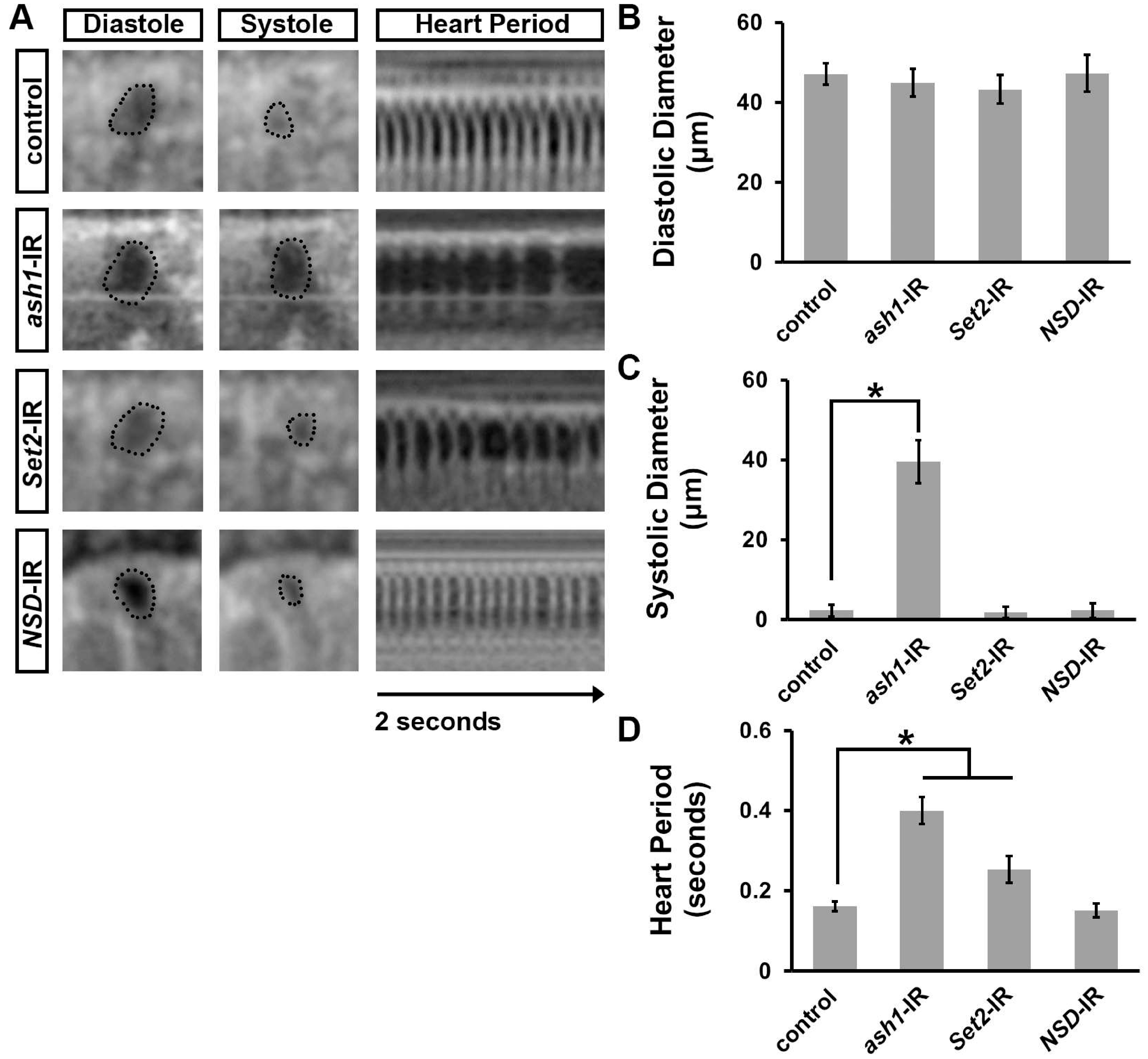
3.1. Silencing ash1 or Set2, Which Encode H3K36 Methyltransferases, Induced Developmental Lethality at the Pupal–Adult Stage
3.2. Silencing ash1 or Set2 Impacted Survival and Induced Cardiac Defects in Adult Drosophila
3.3. Silencing ash1 or Set2 Caused Cardiac Functional Defects in Adult Drosophila
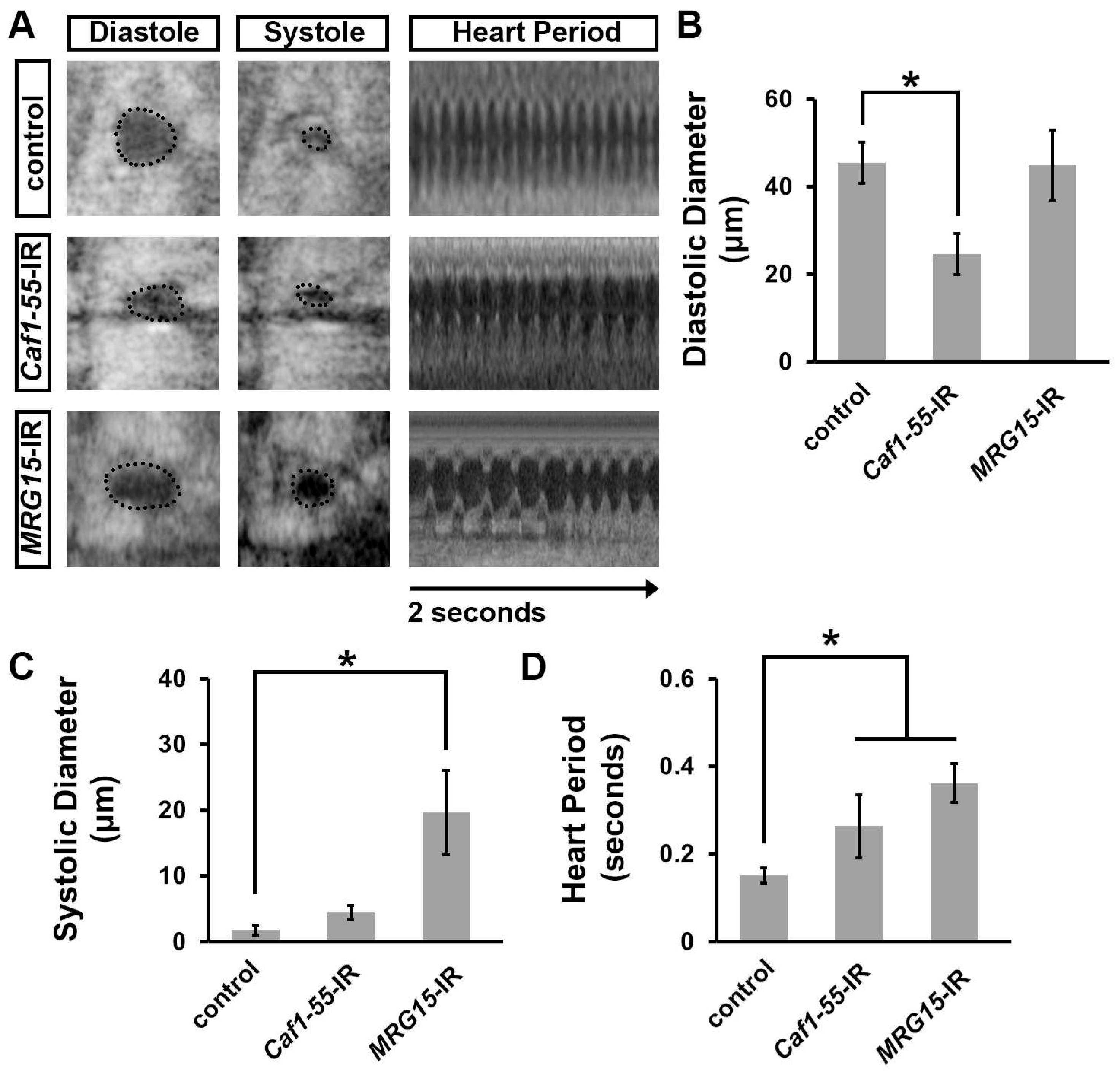
3.4. Silencing Caf1-55 or MRG15, Which Encode Ash1-Complex Components, Increased Developmental Lethality at the Pupal–Adult Stage, Reduced Lifespan, and Caused Heart Morphological Defects
3.5. Silencing Caf1-55 or MRG15 Caused Cardiac Functional Defects in Adult Drosophila
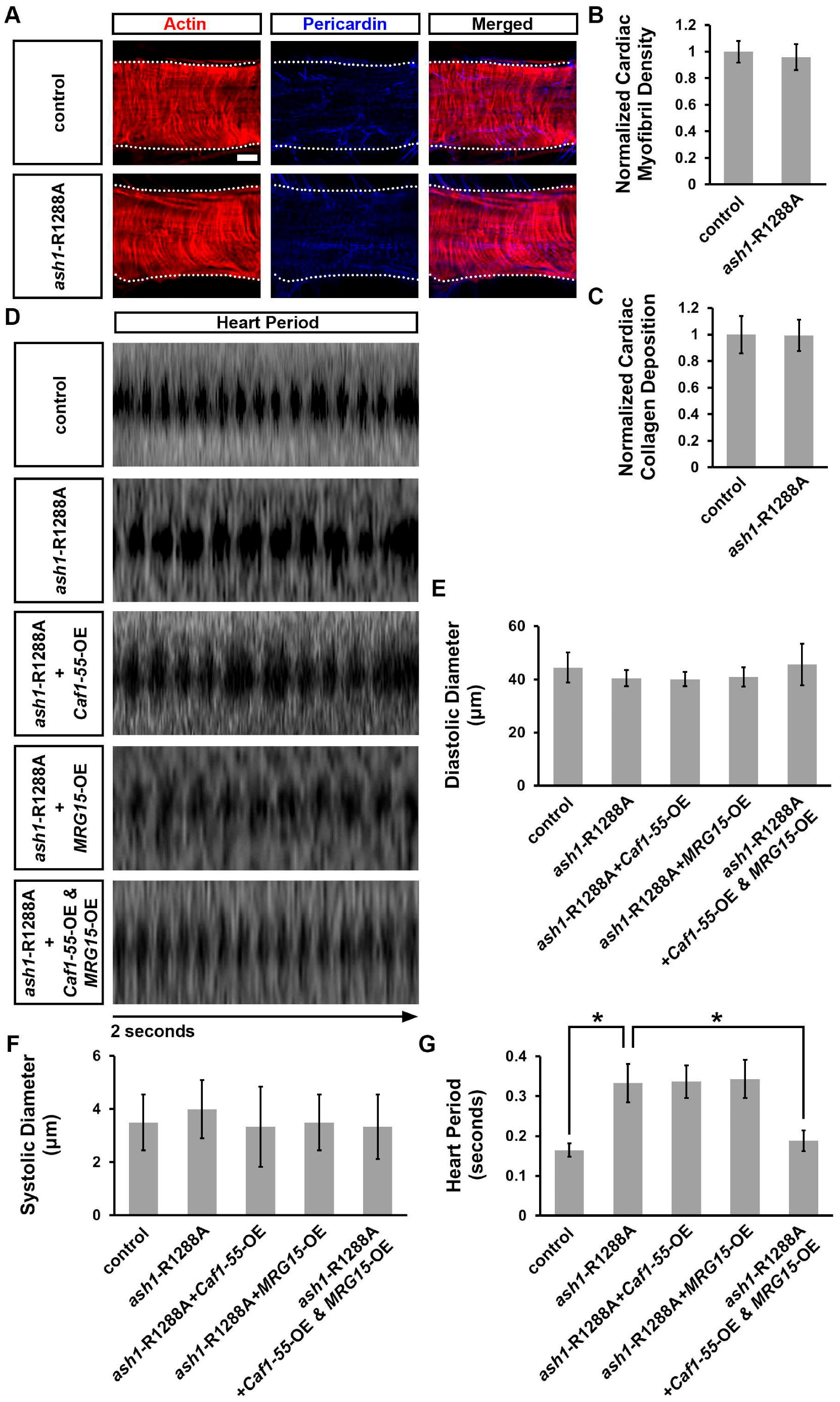
3.6. Simultaneous Overexpression of Caf1-55 and MRG15 Rescued Cardiac Functional Defects Caused by ash1 Mutation
3.7. Simultaneous Overexpression of Caf1-55 and MRG15 Restored the Reduced H3K36me2 Marks Caused by ash1 Mutation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baujat, G.; Cormier-Daire, V. Sotos Syndrome. Orphanet J. Rare Dis. 2007, 2, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergemann, A.D.; Cole, F.; Hirschhorn, K. The Etiology of Wolf-Hirschhorn Syndrome. Trends Genet. 2005, 21, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Cecconi, M.; Forzano, F.; Milani, D.; Cavani, S.; Baldo, C.; Selicorni, A.; Pantaleoni, C.; Silengo, M.; Ferrero, G.; Scarano, G.; et al. Mutation analysis of theNSD1 gene in a group of 59 patients with congenital overgrowth. Am. J. Med. Genet. Part A 2005, 134, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, J.; Wang, H.; Tang, H.; Huang, L.; Wang, S.; Wang, X.; Fang, X.; Liu, J.; Li, L.; et al. Histone Lysine Methyltransferase SETD2 Regulates Coronary Vascular Development in Embryonic Mouse Hearts. Front. Cell Dev. Biol. 2021, 9, 651655. [Google Scholar] [CrossRef]
- Choma, M.A.; Izatt, S.D.; Wessells, R.J.; Bodmer, R.; Izatt, J.A. Images in Cardiovascular Medicine: In Vivo Imaging of the Adult Drosophila Melanogaster Heart with Real-Time Optical Coherence Tomography. Circulation 2006, 114, e35–e36. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.; Azarcon, P.; Hickenlooper, S.; Bia, R.; Horiuchi, E.; Szulik, M.W.; Franklin, S. The Role of Demethylases in Cardiac Development and Disease. J. Mol. Cell. Cardiol. 2021, 158, 89–100. [Google Scholar] [CrossRef]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Huang, X.; Zhang, P.; van de Leemput, J.; Han, Z. Single-cell RNA sequencing identifies novel cell types in Drosophila blood. J. Genet. Genom. 2020, 47, 175–186. [Google Scholar] [CrossRef]
- Gregory, G.D.; Vakoc, C.R.; Rozovskaia, T.; Zheng, X.; Patel, S.; Nakamura, T.; Canaani, E.; Blobel, G.A. Mammalian ASH1L Is a Histone Methyltransferase That Occupies the Transcribed Region of Active Genes. Mol. Cell. Biol. 2007, 27, 8466–8479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, H.P.; Fatima, M.-U.; Pandey, R.; Ram, K.R. Adult exposure of atrazine alone or in combination with carbohydrate diet hastens the onset/progression of type 2 diabetes in Drosophila. Life Sci. 2023, 316, 121370. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Olson, E.N. Hand Is a Direct Target of Tinman and GATA Factors during Drosophila Cardiogenesis and Hematopoiesis. Development 2005, 132, 3525–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Yang, F.; Zhang, Z.; Zhang, J.; Cai, G.; Li, L.; Zheng, Y.; Chen, S.; Xi, R.; Zhu, B. Mrg15 stimulates Ash1 H3K36 methyltransferase activity and facilitates Ash1 Trithorax group protein function in Drosophila. Nat. Commun. 2017, 8, 1649. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Liu, Y.; Wang, Y.; Bailey, C.; Zheng, P.; Liu, Y. Dual Targeting Oncoproteins MYC and HIF1α Regresses Tumor Growth of Lung Cancer and Lymphoma. Cancers 2021, 13, 694. [Google Scholar] [CrossRef]
- Hughes, C.; Turner, S.; Andrews, R.; Vitkin, A.; Jacobs, J. Matrix metalloproteinases regulate ECM accumulation but not larval heart growth in Drosophila melanogaster. J. Mol. Cell. Cardiol. 2020, 140, 42–55. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Ji, W.; Ferdman, D.; Copel, J.; Scheinost, D.; Shabanova, V.; Brueckner, M.; Khokha, M.K.; Ment, L.R. De novo damaging variants associated with congenital heart diseases contribute to the connectome. Sci. Rep. 2020, 10, 7046. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of Rare Inherited and de Novo Variants in 2,871 Congenital Heart Disease Probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.S.; Gelbart, W.M. The Drosophila Polycomb-Group Gene Enhancer of zeste Contains a Region with Sequence Similarity to trithorax. Mol. Cell. Biol. 1993, 13, 6357–6366. [Google Scholar] [CrossRef] [PubMed]
- Larkin, A.; Marygold, S.J.; Antonazzo, G.; Attrill, H.; dos Santos, G.; Garapati, P.V.; Goodman, J.L.; Gramates, L.S.; Millburn, G.; Strelets, V.B.; et al. FlyBase: Updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 2021, 49, D899–D907. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, H.; Song, X.; Song, B.; Liu, W.; Dong, R.; Zhang, H.; Guo, S.; Liang, H.; Schrodi, S.J.; et al. DNA methylome and transcriptome profiling reveal key electrophysiology and immune dysregulation in hypertrophic cardiomyopathy. Epigenetics 2023, 18, 2195307. [Google Scholar] [CrossRef]
- Li, Y.; Trojer, P.; Xu, C.F.; Cheung, P.; Kuo, A.; Drury, W.J., 3rd; Qiao, Q.; Neubert, T.A.; Xu, R.M.; Gozani, O.; et al. The Target of the NSD Family of Histone Lysine Methyltransferases Depends on the Nature of the Substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.W.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 2015, 105, 83–89. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lucio-Eterovic, A.K.; Singh, M.M.; Gardner, J.E.; Veerappan, C.S.; Rice, J.C.; Carpenter, P.B. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc. Natl. Acad. Sci. USA 2010, 107, 16952–16957. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The Human Mitochondrial Transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Petrossian, T.C.; Clarke, S.G. Uncovering the Human Methyltransferasome. Mol. Cell. Proteom. 2011, 10, M110.000976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priest, J.R.; Osoegawa, K.; Mohammed, N.; Nanda, V.; Kundu, R.; Schultz, K.; Lammer, E.J.; Girirajan, S.; Scheetz, T.; Waggott, D.; et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016, 12, e1005963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Zeng, Q.; Wu, H.; Liu, X.; Guida, M.C.; Huang, W.; Zhai, Y.; Li, J.; Ocorr, K.; Bodmer, R.; et al. Deacetylase-Dependent and -Independent Role of HDAC3 in Cardiomyopathy. J. Cell. Physiol. 2023, 238, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Salmand, P.-A.; Iché-Torres, M.; Perrin, L. Tissue-specific cell sorting from Drosophila embryos: Application to gene expression analysis. Fly 2011, 5, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Schmähling, S.; Meiler, A.; Lee, Y.; Mohammed, A.; Finkl, K.; Tauscher, K.; Israel, L.; Borath, M.; Philippou-Massier, J.; Blum, H.; et al. Regulation and function of H3K36 di-methylation by the trithorax-group protein complex AMC. Development 2018, 145, dev163808. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Spletter, M.L.; Barz, C.; Yeroslaviz, A.; Zhang, X.; Lemke, S.B.; Bonnard, A.; Brunner, E.; Cardone, G.; Basler, K.; Habermann, B.H.; et al. A transcriptomics resource reveals a transcriptional transition during ordered sarcomere morphogenesis in flight muscle. Elife 2018, 7, e34058. [Google Scholar] [CrossRef]
- Stassen, M.J.; Bailey, D.; Nelson, S.; Chinwalla, V.; Harte, P.J. The Drosophila trithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mech. Dev. 1995, 52, 209–223. [Google Scholar] [CrossRef]
- Stefanovic, S.; Laforest, B.; Desvignes, J.-P.; Lescroart, F.; Argiro, L.; Maurel-Zaffran, C.; Salgado, D.; Plaindoux, E.; De Bono, C.; Pazur, K.; et al. Hox-dependent coordination of mouse cardiac progenitor cell patterning and differentiation. Elife 2020, 9, e55124. [Google Scholar] [CrossRef]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genom. 2018, 19, 812. [Google Scholar] [CrossRef] [Green Version]
- Szulik, M.W.; Davis, K.; Bakhtina, A.; Azarcon, P.; Bia, R.; Horiuchi, E.; Franklin, S. Transcriptional regulation by methyltransferases and their role in the heart: Highlighting novel emerging functionality. Am. J. Physiol. Circ. Physiol. 2020, 319, H847–H865. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Katagiri, Z.-I.; Kawahashi, K.; Kioussis, D.; Kitajima, S. Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene 2007, 397, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Papadimitriou, J.; Burchell, J.M. Histone Methylases and Demethylases Regulating Antagonistic Methyl Marks: Changes Occurring in Cancer. Cells 2022, 11, 1113. [Google Scholar] [CrossRef]
- Tschiersch, B.; Hofmann, A.; Krauss, V.; Dorn, R.; Korge, G.; Reuter, G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J. 1994, 13, 3822–3831. [Google Scholar] [CrossRef]
- TsNoreau, D.R.; Al-Ata, J.; Jutras, L.; Teebi, A.S. Congenital Heart Defects in Sotos Syndrome. Am. J. Med. Genet. 1999, 84, 172. [Google Scholar]
- Vaughan, L.; Marley, R.; Miellet, S.; Hartley, P.S. The impact of SPARC on age-related cardiac dysfunction and fibrosis in Drosophila. Exp. Gerontol. 2018, 109, 59–66. [Google Scholar] [CrossRef]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Q.; Zhou, Y.; Zhang, S.; Zhang, Y.; Xu, Y.; Yang, Y.; Zhou, C.; Zeng, Z.; Han, J.; Zhu, Q. NSD3, a member of nuclear receptor-binding SET domain family, is a potential prognostic biomarker for pancreatic cancer. Cancer Med. 2023, 12, 10961–10978. [Google Scholar] [CrossRef]
- Yang, K.C.; Yamada, K.A.; Patel, A.Y.; Topkara, V.K.; George, I.; Cheema, F.H.; Ewald, G.A.; Mann, D.L.; Nerbonne, J.M. Deep RNA Sequencing Reveals Dynamic Regulation of Myocardial Noncoding RNAs in Failing Human Heart and Remodeling with Mechanical Circulatory Support. Circulation 2014, 129, 1009–1021. [Google Scholar] [CrossRef]
- Yelbuz, T.M.; Choma, M.A.; Thrane, L.; Kirby, M.L.; Izatt, J.A. Optical Coherence Tomography: A New High-Resolution Imaging Technology to Study Cardiac Development in Chick Embryos. Circulation 2002, 106, 2771–2774. [Google Scholar] [CrossRef] [Green Version]
- Zaghi, M.; Broccoli, V.; Sessa, A. H3K36 Methylation in Neural Development and Associated Diseases. Front. Genet. 2019, 10, 1291. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanoni, P.; Steindl, K.; Sengupta, D.; Joset, P.; Bahr, A.; Sticht, H.; Lang-Muritano, M.; Al, E.; Zweier, M.; Ganzi, O.; et al. Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Di Zhang, D.; Deng, Y.; Kukanja, P.; Agirre, E.; Bartosovic, M.; Dong, M.; Ma, C.; Ma, S.; Su, G.; Bao, S.; et al. Spatial epigenome–transcriptome co-profiling of mammalian tissues. Nature 2023, 616, 113–122. [Google Scholar] [CrossRef]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central Role of Mitofusin 2 in Autophagosome-Lysosome Fusion in Cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.-L.; Zhu, R.-R.; Wu, X.; Xu, H.; Li, Y.-Y.; Xu, Q.-R.; Liu, S.; Huang, H.; Xu, X.; Wan, L.; et al. NSD2 promotes ventricular remodelling mediated by the regulation of H3K36me2. J. Cell. Mol. Med. 2019, 23, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-Y.; Fu, Y.; Nettleton, M.; Richman, A.; Han, Z. High throughput in vivo functional validation of candidate congenital heart disease genes in Drosophila. Elife 2017, 6, e22617. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Fu, Y.; Richman, A.; Han, Z. Validating Candidate Congenital Heart Disease Genes in Drosophila. Bio-Protocol 2017, 7, e2350. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.-y.; Liu, C.; Huang, X.; van de Leemput, J.; Lee, H.; Han, Z. H3K36 Di-Methylation Marks, Mediated by Ash1 in Complex with Caf1-55 and MRG15, Are Required during Drosophila Heart Development. J. Cardiovasc. Dev. Dis. 2023, 10, 307. https://doi.org/10.3390/jcdd10070307
Zhu J-y, Liu C, Huang X, van de Leemput J, Lee H, Han Z. H3K36 Di-Methylation Marks, Mediated by Ash1 in Complex with Caf1-55 and MRG15, Are Required during Drosophila Heart Development. Journal of Cardiovascular Development and Disease. 2023; 10(7):307. https://doi.org/10.3390/jcdd10070307
Chicago/Turabian StyleZhu, Jun-yi, Chen Liu, Xiaohu Huang, Joyce van de Leemput, Hangnoh Lee, and Zhe Han. 2023. "H3K36 Di-Methylation Marks, Mediated by Ash1 in Complex with Caf1-55 and MRG15, Are Required during Drosophila Heart Development" Journal of Cardiovascular Development and Disease 10, no. 7: 307. https://doi.org/10.3390/jcdd10070307
APA StyleZhu, J. -y., Liu, C., Huang, X., van de Leemput, J., Lee, H., & Han, Z. (2023). H3K36 Di-Methylation Marks, Mediated by Ash1 in Complex with Caf1-55 and MRG15, Are Required during Drosophila Heart Development. Journal of Cardiovascular Development and Disease, 10(7), 307. https://doi.org/10.3390/jcdd10070307