
Atrial and Sinoatrial Node Development in the Zebrafish Heart


Abstract
:1. Introduction
2. Mechanisms of Atrial Chamber Development and Chamber Identity Maintenance in the Zebrafish Heart
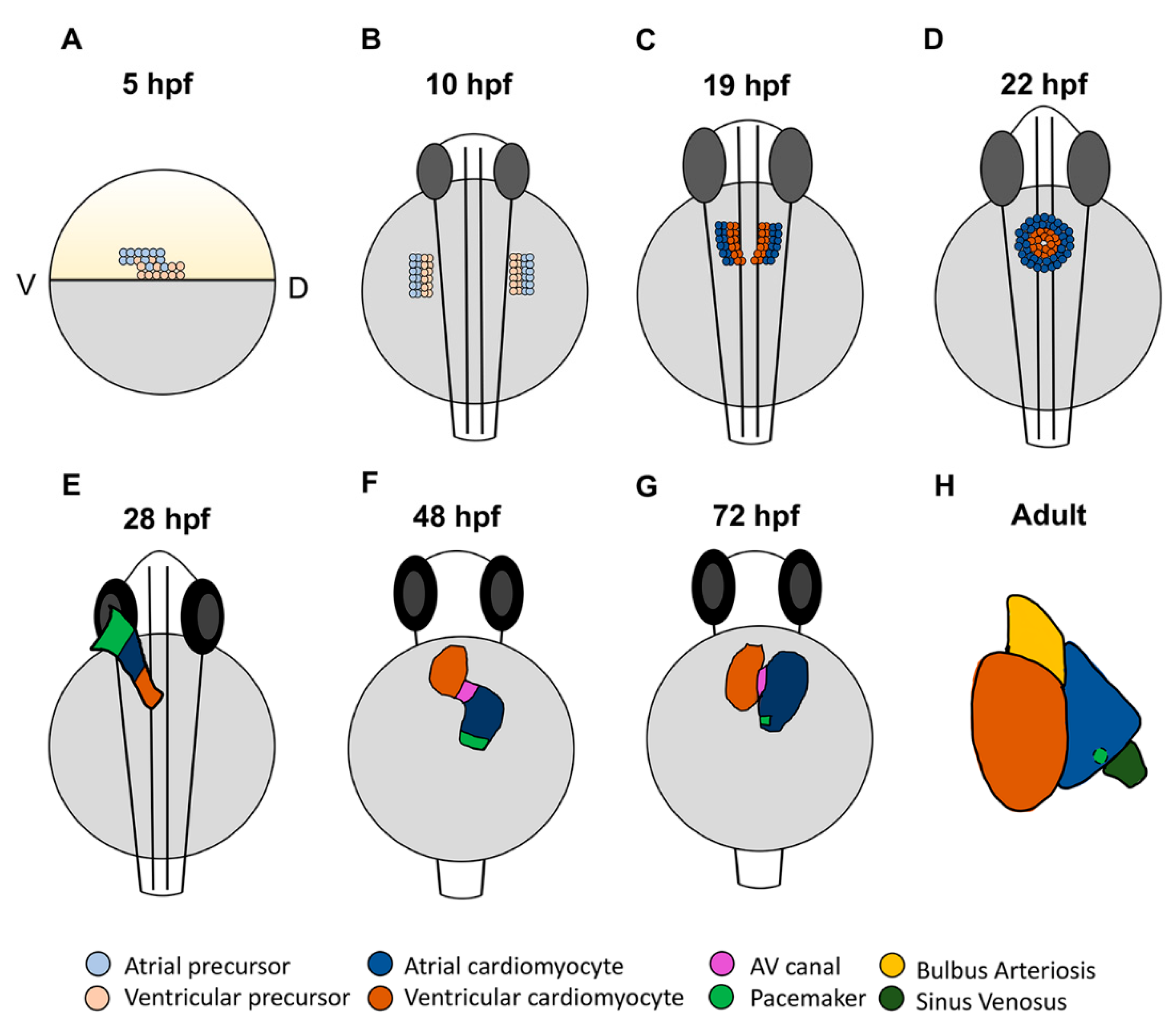
2.1. Cardiac Progenitor Location and Morphology of the Developing Zebrafish Heart
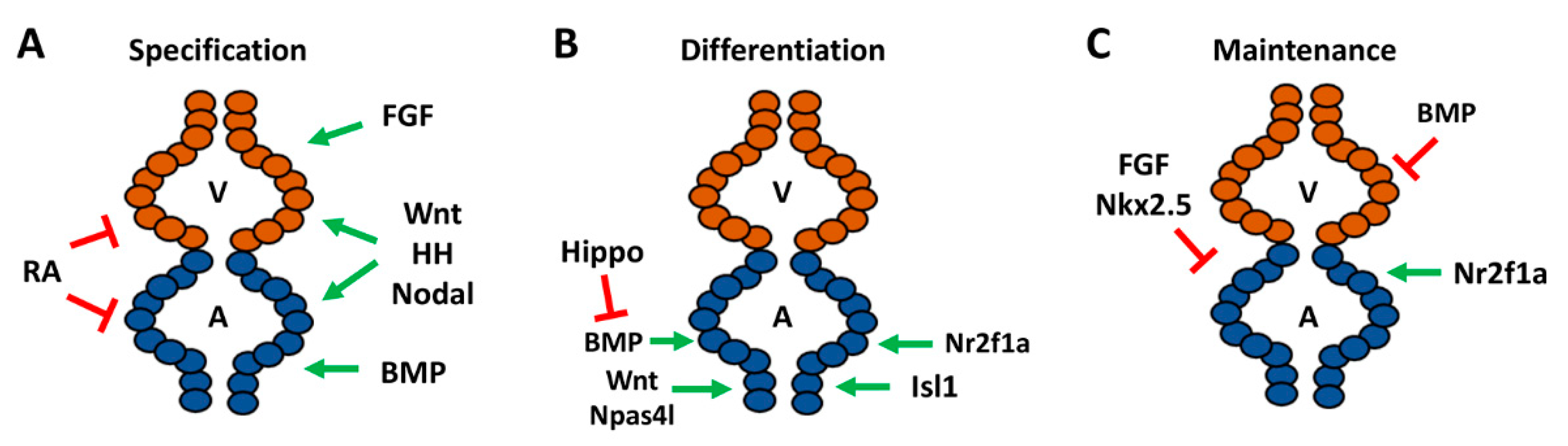
2.2. Signals Regulating the Specification of Atrial Progenitors
2.3. Signals Directing the Differentiation of Atrial Cardiomyocytes
2.4. Maintenance and Plasticity of Cardiomyocyte Identity
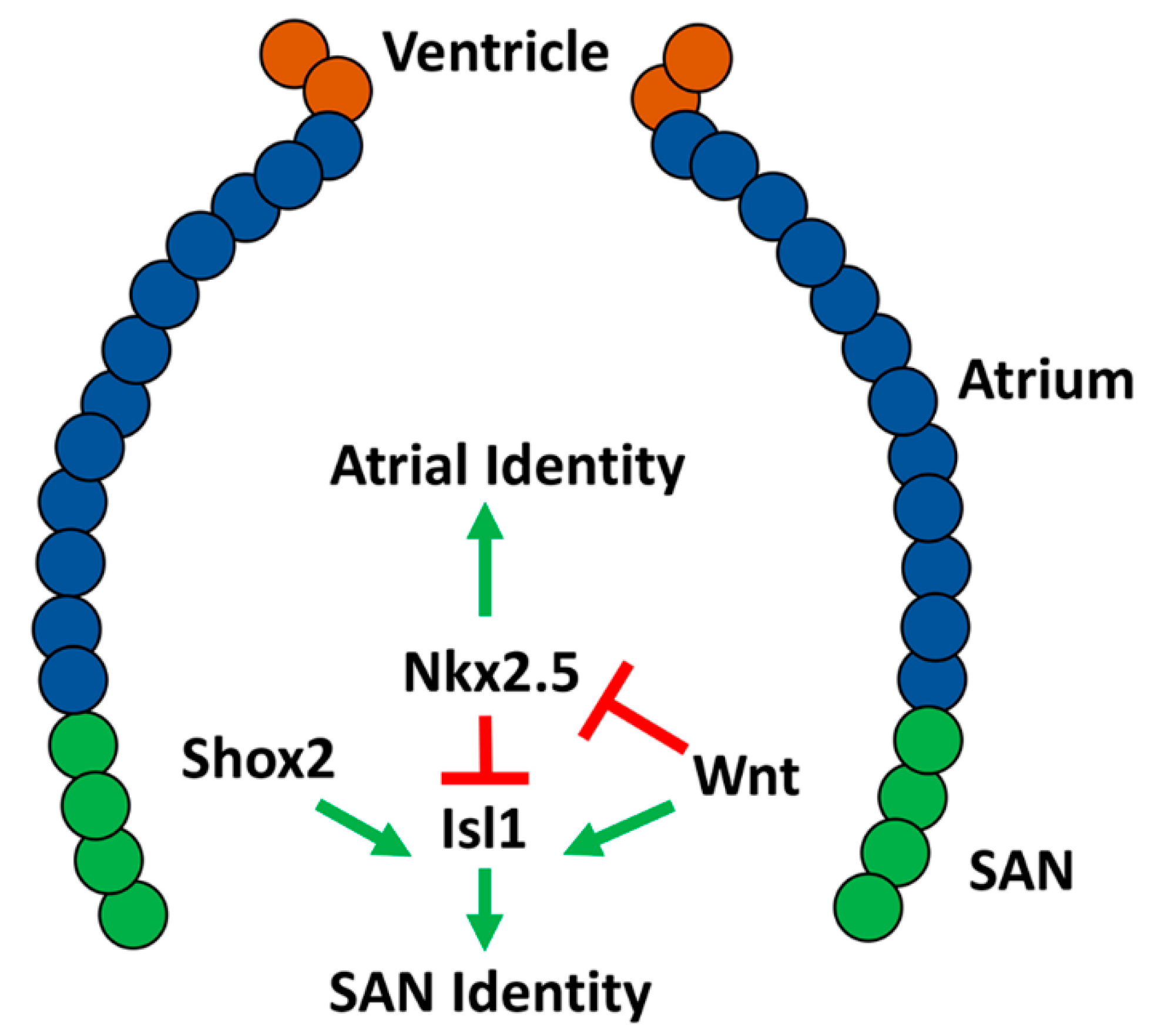
2.5. Conserved Transcriptional Networks Promoting Sinoatrial Node Development
2.6. Development of the Zebrafish Sinoatrial Node
2.7. Zebrafish as a Model for Human Atrial and SAN Defects
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.P.; Lindsay, S.; D’Alessandro, L.C.A.; Swaminathan, G.J.; Bentham, J.; Arndt, A.K.; et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am. J. Hum. Genet. 2014, 94, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, E.; Makita, Y.; Okamoto, T.; Nagaya, K.; Hayashi, T.; Sugimoto, M.; Manabe, H.; Taketazu, G.; Kajino, H.; Fujieda, K. 5.78 Mb terminal deletion of chromosome 15q in a girl, evaluation of NR2F2 as candidate gene for congenital heart defects. Eur. J. Med. Genet. 2011, 54, 354–356. [Google Scholar] [CrossRef]
- Benson, D.W.; Silberbach, G.M.; Kavanaugh-McHugh, A.; Cottrill, C.; Zhang, Y.; Riggs, S.; Smalls, O.; Johnson, M.C.; Watson, M.S.; Seidman, J.G.; et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J. Clin. Investig. 1999, 104, 1567–1573. [Google Scholar] [CrossRef] [Green Version]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef]
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, C.A. Epidemiology of cardiovascular malformations: Prevalence and risk factors. Am. J. Med. Genet. 2000, 97, 319–325. [Google Scholar] [CrossRef]
- Van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics-2018 update: A report from the American Heart Association. Circulation 2018, 137, E67–E492. [Google Scholar] [CrossRef] [PubMed]
- Marelli, A.J.; Mackie, A.S.; Ionescu-Ittu, R.; Rahme, E.; Pilote, L. Congenital heart disease in the general population: Changing prevalence and age distribution. Circulation 2007, 115, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G.; Logan, M.; Davis, N.; Levi, T.; Tabin, C.J.; Seidman, J.G.; Seidman, C.E. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt- Oram syndrome. Dev. Biol. 1999, 211, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, R.M.; Kumar, S. Sinus node and atrial arrhythmias. Circulation 2016, 133, 1892–1900. [Google Scholar] [CrossRef]
- Choudhury, M.; Boyett, M.R.; Morris, G.M. Biology of the sinus node and its disease. Arrhythmia Electrophysiol. Rev. 2015, 4, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Paone, C.; Sumer, S.A.; Diebold, S.; Weiss, B.; Roeth, R.; Clauss, S.; Klier, I.; Kääb, S.; Schulz, A.; et al. Functional characterization of rare variants in the SHOX2 gene identified in sinus node dysfunction and atrial fibrillation. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 2006, 354, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, S.; Aziz, P.F.; Saarel, E. Expanding the electrical phenotype of NKX2-5 mutations: Ventricular tachycardia, atrial fibrillation, and complete heart block within one family. Hear. Case Rep. 2018, 4, 530–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vornanen, M.; Hassinen, M. Zebrafish heart as a model for human cardiac electrophysiology. Channels 2016, 10, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardoglou, P.; Beis, D. On zebrafish disease models and matters of the heart. Biomedicines 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Samsa, L.; Qian, L.; Liu, J. Advances in the study of heart development and disease using zebrafish. J. Cardiovasc. Dev. Dis. 2016, 3, 13. [Google Scholar] [CrossRef]
- Poon, K.L.; Brand, T. The zebrafish model system in cardiovascular research: A tiny fish with mighty prospects. Glob. Cardiol. Sci. Pract. 2013, 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravens, U. Ionic basis of cardiac electrophysiology in zebrafish compared to human hearts. Prog. Biophys. Mol. Biol. 2018, 138, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Chi, N.C.; Shaw, R.M.; Jungblut, B.; Huisken, J.; Ferrer, T.; Arnaout, R.; Scott, I.; Beis, D.; Xiao, T.; Baier, H.; et al. Genetic and physiologic dissection of the vertebrate cardiac conduction system. PLoS Biol. 2008, 6, 1006–1019. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, S.; Kohl, T.; Williams, G.S.B.; Gusev, K.; Wagner, E.; Rog-Zielinska, E.A.; Hebisch, E.; Dura, M.; Didié, M.; Gotthardt, M.; et al. Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Investig. 2016, 126, 3999–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyrnias, I.; Mair, W.; Harzheim, D.; Walker, S.A.; Roderick, H.L.; Bootman, M.D. Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium 2010, 47, 210–223. [Google Scholar] [CrossRef]
- Bloomekatz, J.; Galvez-Santisteban, M.; Chi, N.C. Myocardial plasticity: Cardiac development, regeneration and disease. Curr. Opin. Genet. Dev. 2016, 40, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Tessadori, F.; van Weerd, J.H.; Burkhard, S.B.; Verkerk, A.O.; de Pater, E.; Boukens, B.J.; Vink, A.; Christoffels, V.M.; Bakkers, J. Identification and functional characterization of cardiac pacemaker cells in zebrafish. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabibiazar, R.; Wagner, R.A.; Liao, A.; Quertermous, T. Transcriptional profiling of the heart reveals chamber-specific gene expression patterns. Circ. Res. 2003, 93, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Wong, C.K.; Tsang, S.Y. Differential gene expressions in atrial and ventricular myocytes: Insights into the road of applying embryonic stem cell-derived cardiomyocytes for future therapies. Am. J. Physiol. Physiol. 2010, 299, C1234–C1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootman, M.D.; Higazi, D.R.; Coombes, S.; Roderick, H.L. Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J. Cell Sci. 2006, 119, 3915–3925. [Google Scholar] [CrossRef] [Green Version]
- Nof, E.; Antzelevitch, C.; Glikson, M. The contribution of HCN4 to normal sinus node function in humans and animal models. Pacing Clin. Electrophysiol. 2010, 33, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Christoffels, V.M.; Smits, G.J.; Kispert, A.; Moorman, A.F.M. Development of the pacemaker tissues of the heart. Circ. Res. 2010, 106, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.; Lee, R.K.; Fishman, M.C. Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development 1993, 119, 31–40. [Google Scholar] [PubMed]
- Berdougo, E.; Coleman, H.; Lee, D.H.; Stainier, D.Y.R.; Yelon, D. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development 2003, 130, 6121–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudt, D.; Stainier, D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annu. Rev. Genet. 2012, 46, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Keegan, B.R.; Meyer, D.; Yelon, D. Organization of cardiac chamber progenitors in the zebrafish blastula. Development 2004, 131, 3081–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelon, D.; Stainier, D.Y.R. Patterning during organogenesis: Genetic analysis of cardiac chamber formation. Semin. Cell Dev. Biol. 1999, 10, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, A.; Germano, R.F.V.; Stone, O.; Arnaout, R.; Guenther, S.; Ahuja, S.; Uribe, V.; Vanhollebeke, B.; Stainier, D.Y.R.; Reischauer, S. Distinct myocardial lineages break atrial symmetry during cardiogenesis in zebrafish. Elife 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Tessari, A.; Pietrobon, M.; Notte, A.; Cifelli, G.; Gage, P.J.; Schneider, M.D.; Lembo, G.; Campione, M. Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ. Res. 2008, 102, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Rohr, S.; Otten, C.; Abdelilah-Seyfried, S. Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ. Res. 2008, 102. [Google Scholar] [CrossRef] [Green Version]
- Yelon, D.; Horne, S.A.; Stainier, D.Y.R. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev. Biol. 1999, 214, 23–37. [Google Scholar] [CrossRef] [Green Version]
- De Pater, E.; Clijsters, L.; Marques, S.R.; Lin, Y.F.; Garavito-Aguilar, Z.V.; Yelon, D.; Bakkers, J. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development 2009, 136, 1633–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Stainier, D.Y.R. Zebrafish in the study of early cardiac development. Circ. Res. 2012, 110, 870–874. [Google Scholar] [CrossRef]
- Hami, D.; Grimes, A.C.; Tsai, H.J.; Kirby, M.L. Zebrafish cardiac development requires a conserved secondary heart field. Development 2011, 138, 2389–2398. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Lazic, S.; Scott, I.C. Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev. Biol. 2011, 354, 123–133. [Google Scholar] [CrossRef]
- Hu, N.; Sedmera, D.; Yost, H.J.; Clark, E.B. Structure and function of the developing zebrafish heart. Anat. Rec. 2000, 260, 148–157. [Google Scholar] [CrossRef]
- Gupta, V.; Poss, K.D. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 2012, 484, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Bressan, M.; Hassel, D.; Huisken, J.; Staudt, D.; Kikuchi, K.; Poss, K.D.; Mikawa, T.; Stainier, D.Y.R. A dual role for ErbB2 signaling in cardiac trabeculation. Development 2010, 137, 3867–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foglia, M.J.; Cao, J.; Tornini, V.A.; Poss, K.D. Multicolor mapping of the cardiomyocyte proliferation dynamics that construct the atrium. Development 2016, 143, 1688–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.A.; Koudijs, M.; van Eeden, F.J.M.; Joyner, A.L.; Yelon, D. Hedgehog signaling plays a cell-autonomous role in maximizing cardiac developmental potential. Development 2008, 135, 3789–3799. [Google Scholar] [CrossRef] [Green Version]
- Dohn, T.E.; Waxman, J.S. Distinct phases of Wnt/β-catenin signaling direct cardiomyocyte formation in zebrafish. Dev. Biol. 2012, 361, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Ueno, S.; Weidinger, G.; Osugi, T.; Kohn, A.D.; Golob, J.L.; Pabon, L.; Reinecke, H.; Moon, R.T.; Murry, C.E. Biphasic role for Wnt/β-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9685–9690. [Google Scholar] [CrossRef] [Green Version]
- Deshwar, A.R.; Chng, S.C.; Ho, L.; Reversade, B.; Scott, I.C. The Apelin receptor enhances Nodal/TGFβ signaling to ensure proper cardiac development. Elife 2016, 5. [Google Scholar] [CrossRef]
- Scott, I.C.; Masri, B.; D’Amico, L.A.; Jin, S.W.; Jungblut, B.; Wehman, A.M.; Baier, H.; Audigier, Y.; Stainier, D.Y.R. The G Protein-Coupled receptor agtrl1b regulates early development of myocardial progenitors. Dev. Cell 2007, 12, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Fishman, M.C. Patterning the zebrafish heart tube: Acquisition of anteroposterior polarity. Dev. Biol. 1992, 153, 91–101. [Google Scholar] [CrossRef]
- Hochgreb, T.; Linhares, V.L.; Menezes, D.C.; Sampaio, A.C.; Yan, C.Y.I.; Cardoso, W.V.; Rosenthal, N.; Xavier-Neto, J. A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 2003, 130, 5363–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, B.R.; Feldman, J.L.; Begemann, G.; Ingham, P.W.; Yelon, D. Retinoic acid signaling restricts the cardiac progenitor pool. Science 2005, 307, 247–249. [Google Scholar] [CrossRef]
- Waxman, J.S.; Keegan, B.R.; Roberts, R.W.; Poss, K.D.; Yelon, D. Hoxb5b Acts Downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 2008, 15, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Waxman, J.S.; Yelon, D. Increased hox activity mimics the teratogenic effects of excess retinoic acid signaling. Dev. Dyn. 2009, 238, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- D’Aniello, E.; Rydeen, A.B.; Anderson, J.L.; Mandal, A.; Waxman, J.S. Depletion of retinoic acid receptors initiates a novel positive feedback mechanism that promotes teratogenic increases in retinoic acid. PLoS Genet. 2013, 9, 1003689. [Google Scholar] [CrossRef] [Green Version]
- Marques, S.R.; Lee, Y.; Poss, K.D.; Yelon, D. Reiterative roles for FGF signaling in the establishment of size and proportion of the zebrafish heart. Dev. Biol. 2008, 321, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Marques, S.R.; Yelon, D. Differential requirement for BMP signaling in atrial and ventricular lineages establishes cardiac chamber proportionality. Dev. Biol. 2009, 328, 472–482. [Google Scholar] [CrossRef] [Green Version]
- De Pater, E.; Ciampricotti, M.; Priller, F.; Veerkamp, J.; Strate, I.; Smith, K.; Lagendijk, A.K.; Schilling, T.F.; Herzog, W.; Abdelilah-Seyfried, S.; et al. Bmp signaling exerts opposite effects on cardiac differentiation. Circ. Res. 2012, 110, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Reischauer, S.; Stone, O.A.; Villasenor, A.; Chi, N.; Jin, S.W.; Martin, M.; Lee, M.T.; Fukuda, N.; Marass, M.; Witty, A.; et al. Cloche is a bHLH-PAS transcription factor that drives haemato-vascular specification. Nature 2016, 535, 294–298. [Google Scholar] [CrossRef]
- Schoenebeck, J.J.; Keegan, B.R.; Yelon, D. Vessel and blood specification override cardiac potential in anterior mesoderm. Dev. Cell 2007, 13, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.-L. The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 2015, 116, 1431–1447. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Miyazaki, T.; Chow, R.W.Y.; Ishikawa, H.; Nakajima, H.; Vermot, J.; Mochizuki, N. Hippo signaling determines the number of venous pole cells that originate from the anterior lateral plate mesoderm in zebrafish. Elife 2018, 7. [Google Scholar] [CrossRef]
- Witzel, H.R.; Cheedipudi, S.; Gao, R.; Stainier, D.Y.R.; Dobreva, G.D. Isl2b regulates anterior second heart field development in zebrafish. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Pereira, F.A.; Yuhong, Q.; Zhou, G.; Tsai, M.J.; Tsai, S.Y. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. 1999, 13, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, T.B.; Ravisankar, P.; Song, Y.C.; Gafranek, J.T.; Rydeen, A.B.; Dohn, T.E.; Barske, L.A.; Crump, J.G.; Waxman, J.S. Nr2f1a balances atrial chamber and atrioventricular canal size via BMP signaling-independent and -dependent mechanisms. Dev. Biol. 2018, 434, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.S.; Merk, S.; Arnoldi, E.; Zwermann, L.; Kloos, P.; Gebauer, M.; Steinmeyer, K.; Bleich, M.; Kääb, S.; Pfeufer, A.; et al. Functional profiling of human atrial and ventricular gene expression. Pflugers Arch. Eur. J. Physiol. 2005, 450, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Small, E.M.; Van Rooij, E.; Qi, X.; Richardson, J.A.; Srivastava, D.; Nakagawa, O.; Olson, E.N. Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc. Natl. Acad. Sci. USA 2007, 104, 7975–7980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targoff, K.L.; Colombo, S.; George, V.; Schell, T.; Kim, S.H.; Solnica-Krezel, L.; Yelon, D. Nkx genes are essential for maintenance of ventricular identity. Development 2013, 140, 4203–4213. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.; Zeng, X.X.I.; Sidhwani, P.; Marques, S.R.; George, V.; Targoff, K.L.; Chi, N.C.; Yelon, D. FGF signaling enforces cardiac chamber identity in the developing ventricle. Development 2017, 144, 1328–1338. [Google Scholar] [CrossRef] [Green Version]
- George, V.; Colombo, S.; Targoff, K.L. An early requirement for nkx2.5 ensures the first and second heart field ventricular identity and cardiac function into adulthood. Dev. Biol. 2015, 400, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifers, F.; Walsh, E.C.; Léger, S.; Stainier, D.Y.R.; Brand, M. Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar). Development 2000, 127, 225–235. [Google Scholar]
- Zhang, R.; Han, P.; Yang, H.; Ouyang, K.; Lee, D.; Lin, Y.F.; Ocorr, K.; Kang, G.; Chen, J.; Stainier, D.Y.R.; et al. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 2013, 498, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.P.; Cheng, C.M.; Lanz, R.B.; Wang, T.; Respress, J.L.; Ather, S.; Chen, W.; Tsai, S.J.; Wehrens, X.H.T.; Tsai, M.J.; et al. Atrial identity is determined by a COUP-TFII regulatory network. Dev. Cell 2013, 25, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrenberg, A.B.; Stainier, Y.R.; Baier, H.; Huisken, J. Optogenetic control of cardiac function. Source Sci. 2010, 330, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Burkhard, S.; van Eif, V.; Garric, L.; Christoffels, V.; Bakkers, J. On the evolution of the cardiac pacemaker. J. Cardiovasc. Dev. Dis. 2017, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Wiese, C.; Grieskamp, T.; Airik, R.; Mommersteeg, M.T.M.; Gardiwal, A.; De Gier-De Vries, C.; Schuster-Gossler, K.; Moorman, A.F.M.; Kispert, A.; Christoffels, V.M. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res. 2009, 104, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Hoogaars, W.M.H.; Engel, A.; Brons, J.F.; Verkerk, A.O.; De Lange, F.J.; Wong, L.Y.E.; Bakker, M.L.; Clout, D.E.; Wakker, V.; Barnett, P.; et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007, 21, 1098–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza-Lewis, R.A.; Liu, H.; Sun, C.; Chen, C.; Jiao, K.; Chen, Y.P. Ectopic expression of Nkx2.5 suppresses the formation of the sinoatrial node in mice. Dev. Biol. 2011, 356, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, Y.; Yanez, D.A.; Touma, M.; Nakano, H.; Jaroszewicz, A.; Jordan, M.C.; Pellegrini, M.; Roos, K.P.; Nakano, A. Nkx2-5 suppresses the proliferation of atrial myocytes and conduction system. Circ. Res. 2014, 114, 1103–1113. [Google Scholar] [CrossRef]
- Mommersteeg, M.T.M.; Hoogaars, W.M.H.; Prall, O.W.J.; De Gier-De Vries, C.; Wiese, C.; Clout, D.E.W.; Papaioannou, V.E.; Brown, N.A.; Harvey, R.P.; Moorman, A.F.M.; et al. Molecular pathway for the localized formation of the sinoatrial node. Circ. Res. 2007, 100, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liang, X.; Najafi, N.; Cass, M.; Lin, L.; Cai, C.L.; Chen, J.; Evans, S.M. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev. Biol. 2007, 304, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, F.; Mehrkens, D.; Friedrich, F.W.; Stubbendorff, M.; Hua, X.; Müller, J.C.; Schrepfer, S.; Evans, S.M.; Carrier, L.; Eschenhagen, T. Localization of islet-1-positive cells in the healthy and infarcted adult murine heart. Circ. Res. 2012, 110, 1303–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaschke, R.J.; Hahurij, N.D.; Kuijper, S.; Just, S.; Wisse, L.J.; Deissler, K.; Maxelon, T.; Anastassiadis, K.; Spitzer, J.; Hardt, S.E.; et al. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation 2007, 115, 1830–1838. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Zhang, Q.; Cattaneo, P.; Zhuang, S.; Gong, X.; Spann, N.J.; Jiang, C.; Cao, X.; Zhao, X.; Zhang, X.; et al. Transcription factor ISL1 is essential for pacemaker development and function. J. Clin. Investig. 2015, 125, 3256–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza-Lewis, R.A.; Yu, L.; He, F.; Liu, H.; Tang, R.; Shi, J.; Sun, X.; Martin, J.F.; Wang, D.; Yang, J.; et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev. Biol. 2009, 327, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, C.H.; Espinoza-Lewis, R.A.; Jiao, Z.; Sheu, I.; Hu, X.; Lin, M.; Zhang, Y.; Chen, Y.P. Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J. Biol. Chem. 2011, 286, 17029–17038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhard, S.B.; Bakkers, J. Spatially resolved RNA-sequencing of the embryonic heart identifies a role for Wnt/β-catenin signaling in autonomic control of heart rate. Elife 2018, 7. [Google Scholar] [CrossRef]
- Hoffmann, S.; Berger, I.M.; Glaser, A.; Bacon, C.; Li, L.; Gretz, N.; Steinbeisser, H.; Rottbauer, W.; Just, S.; Rappold, G. Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res. Cardiol. 2013, 108, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, S.; De Sena-Tomaś, C.; George, V.; Werdich, A.A.; Kapur, S.; Macrae, C.A.; Targoff, K.L. Nkx genes establish second heart field cardiomyocyte progenitors at the arterial pole and pattern the venous pole through isl1 repression. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressan, M.; Liu, G.; Mikawa, T. Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science 2013, 340, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Mommersteeg, M.T.M.; Domínguez, J.N.; Wiese, C.; Norden, J.; De Gier-De Vries, C.; Burch, J.B.E.; Kispert, A.; Brown, N.A.; Moorman, A.F.M.; Christoffels, V.M. The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc. Res. 2010, 87, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Han, P.; Ma, X.; Farah, E.N.; Bloomekatz, J.; Zeng, X.X.I.; Zhang, R.; Swim, M.M.; Witty, A.D.; Knight, H.G.; et al. Canonical Wnt5b Signaling Directs Outlying Nkx2.5+ Mesoderm into Pacemaker Cardiomyocytes. Dev. Cell 2019, 50, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Han, P.; Kim, E.H.; Mak, J.; Zhang, R.; Torrente, A.G.; Goldhaber, J.I.; Marbán, E.; Cho, H.C. Canonical Wnt signaling promotes pacemaker cell specification of cardiac mesodermal cells derived from mouse and human embryonic stem cells. Stem Cells 2020, 38, 352–368. [Google Scholar] [CrossRef]
- Suradi, H.S.; Hijazi, Z.M. Adult congenital interventions in heart failure. Interv. Cardiol. Clin. 2017, 6, 427–443. [Google Scholar] [CrossRef]
- Rodriguez, F.H.; Moodie, D.S.; Parekh, D.R.; Franklin, W.J.; Morales, D.L.S.; Zafar, F.; Graves, D.E.; Friedman, R.A.; Rossano, J.W. Outcomes of hospitalization in adults in the United States with atrial septal defect, ventricular septal defect, and atrioventricular septal defect. Am. J. Cardiol. 2011, 108, 290–293. [Google Scholar] [CrossRef]
- Webb, G.; Gatzoulis, M.A. Atrial septal defects in the adult: Recent progress and overview. Circulation 2006, 114, 1645–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, M.B.; Wilmanns, J.C.; Chandran, A.; Perera, J.; Hon, O.; Biben, C.; Willow, T.J.; Nim, H.T.; Kaur, G.; Simonds, S.; et al. Point mutations in murine Nkx2-5 phenocopy human congenital heart disease and induce pathogenic Wnt signaling. JCI Insight 2017, 2, e88271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Bahr, E.; Neu, A.; Friederich, P.; Kaupp, U.B.; Breithardt, G.; Pongs, O.; Isbrandt, D. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Investig. 2003, 111, 1537–1545. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Clauss, S.; Berger, I.M.; Weiß, B.; Montalbano, A.; Röth, R.; Bucher, M.; Klier, I.; Wakili, R.; Seitz, H.; et al. Coding and non-coding variants in the SHOX2 gene in patients with early-onset atrial fibrillation. Basic Res. Cardiol. 2016, 111, 36. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, Z.S.; Wang, X.H.; Xu, Y.J.; Qiao, Q.; Li, X.M.; Di, R.M.; Guo, X.J.; Li, R.G.; Zhang, M.; et al. A SHOX2 loss-of-function mutation underlying familial atrial fibrillation. Int. J. Med. Sci. 2018, 15, 1564–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.R.; Perry, J.C. Arrhythmias and conduction disorders associated with atrial septal defects. J. Thorac. Dis. 2018, 10, S2940–S2944. [Google Scholar] [CrossRef] [PubMed]
- Ellesøe, S.G.; Johansen, M.M.; Bjerre, J.V.; Hjortdal, V.E.; Brunak, S.; Larsen, L.A. Familial atrial septal defect and sudden cardiac death: Identification of a novel NKX2-5 mutation and a review of the literature. Congenit. Heart Dis. 2016, 11, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillutla, P.; Shetty, K.D.; Foster, E. Mortality associated with adult congenital heart disease: Trends in the US population from 1979 to 2005. Am. Heart J. 2009, 158, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Hoffmann, A.D.; Burnicka-Turek, O.; Friedland-Little, J.M.; Zhang, K.; Moskowitz, I.P. Tbx5-Hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.L.; Sribudiani, Y.; Dong, X.; Huber, C.; Kois, C.; Baujat, G.; Gordon, C.T.; Mayne, V.; Galmiche, L.; Serre, V.; et al. Bi-allelic variations of SMO in humans cause a broad spectrum of developmental anomalies due to abnormal hedgehog signaling. Am. J. Hum. Genet. 2020, 106, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.M.; Rho, S.; Petiet, A.; Davenport, C.L.; Johnson, G.A.; Meyers, E.N.; Klingensmith, J.; Goddeeris, M.M. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoak, I.W.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Garrity, D.M.; Childs, S.; Fishman, M.C. The heartstrings mutation in zebrafish causes heart/fin Tbx5 deficiency syndrome. Development 2002, 129, 4635–4645. [Google Scholar]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef]
- Sirbu, I.O.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn. 2008, 237, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [Green Version]
- Dohn, T.E.; Ravisankar, P.; Tirera, F.T.; Martin, K.E.; Gafranek, J.T.; Duong, T.B.; van Dyke, T.L.; Touvron, M.; Barske, L.A.; Crump, J.G.; et al. Nr2f-dependent allocation of ventricular cardiomyocyte and pharyngeal muscle progenitors. PLoS Genet. 2019, 15. [Google Scholar] [CrossRef]
- Qiao, X.H.; Wang, Q.; Wang, J.; Liu, X.Y.; Xu, Y.J.; Huang, R.T.; Xue, S.; Li, Y.J.; Zhang, M.; Qu, X.K.; et al. A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur. J. Med. Genet. 2018, 61, 197–203. [Google Scholar] [CrossRef]
- Upadia, J.; Gonzales, P.R.; Robin, N.H. Novel de novo pathogenic variant in the NR2F2 gene in a boy with congenital heart defect and dysmorphic features. Am. J. Med. Genet. Part A 2018, 176, 1423–1426. [Google Scholar] [CrossRef]
- Elliott, D.A.; Kirk, E.P.; Yeoh, T.; Chandar, S.; McKenzie, F.; Taylor, P.; Grossfeld, P.; Fatkin, D.; Jones, O.; Hayes, P.; et al. Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: Associations with atrial septal defect and hypoplastic left heart syndrome. J. Am. Coll. Cardiol. 2003, 41, 2072–2076. [Google Scholar] [CrossRef] [Green Version]
- Harrington, J.K.; Sorabella, R.; Tercek, A.; Isler, J.R.; Targoff, K.L. Nkx2.5 is essential to establish normal heart rate variability in the zebrafish embryo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R265–R271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElhinney, D.B.; Geiger, E.; Blinder, J.; Benson, D.W.; Goldmuntz, E. NKX2.5 Mutations in patients with congenital heart disease. J. Am. Coll. Cardiol. 2003, 42, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targoff, K.L.; Schell, T.; Yelon, D. Nkx genes regulate heart tube extension and exert differential effects on ventricular and atrial cell number. Dev. Biol. 2008, 322, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatami, M.; Heidari, M.M.; Kazeminasab, F.; Zare Bidaki, R. Identification of a novel non-sense mutation in TBX5 gene in pediatric patients with congenital heart defects. J. Cardiovasc. Thorac. Res. 2018, 10, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Pi-Roig, A.; Martin-Blanco, E.; Minguillon, C. Distinct tissue-specific requirements for the zebrafish tbx5 genes during heart, retina and pectoral fin development. Open Biol. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Jou, C.J.; Arrington, C.B.; Barnett, S.; Shen, J.; Cho, S.; Sheng, X.; McCullagh, P.C.; Bowles, N.E.; Pribble, C.M.; Saarel, E.V.; et al. A functional assay for sick sinus syndrome genetic variants. Cell. Physiol. Biochem. 2017, 42, 2021–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Poot, M.; Eleveld, M.J.; van ’t Slot, R.; van Genderen, M.M.; Verrijn Stuart, A.A.; Hochstenbach, R.; Beemer, F.A. Proportional growth failure and oculocutaneous albinism in a girl with a 6.87 Mb deletion of region 15q26.2→qter. Eur. J. Med. Genet. 2007, 50, 432–440. [Google Scholar] [CrossRef]
- Brown, K.K.; Alkuraya, F.S.; Matos, M.; Robertson, R.L.; Kimonis, V.E.; Morton, C.C. NR2F1 deletion in a patient with a de novo paracentric inversion, inv(5)(q15q33.2), and syndromic deafness. Am. J. Med. Genet. Part A 2009, 149, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Quaranta, R.; Fell, J.; Rühle, F.; Rao, J.; Piccini, I.; Araúzo-Bravo, M.J.; Verkerk, A.O.; Stoll, M.; Greber, B. Revised roles of ISL1 in a hES cell-based model of human heart chamber specification. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Xu, J.H.; Song, H.M.; Zhao, L.; Xu, W.J.; Wang, J.; Li, R.G.; Xu, L.; Jiang, W.F.; Qiu, X.B.; et al. Mutational spectrum of the NKX2-5 gene in patients with lone atrial fibrillation. Int. J. Med. Sci. 2014, 11, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.H.; Chang, C.; Xu, Y.J.; Li, R.G.; Qu, X.K.; Fang, W.Y.; Liu, X.; Yang, Y.Q. Prevalence and spectrum of Nkx2.5 mutations associated with idiopathic atrial fibrillation. Clinics 2013, 68, 777–784. [Google Scholar] [CrossRef]
- Lyons, I.; Parsons, L.M.; Hartley, L.; Li, R.; Andrews, J.E.; Robb, L.; Harvey, R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995, 9, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Chen, Z.; Bartunkova, S.; Yamasaki, N.; Izumo, S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999, 126, 1269–1280. [Google Scholar] [PubMed]
- Ashraf, H.; Pradhan, L.; Chang, E.I.; Terada, R.; Ryan, N.J.; Briggs, L.E.; Chowdhury, R.; Zárate, M.A.; Sugi, Y.; Nam, H.J.; et al. A mouse model of human congenital heart disease high incidence of diverse cardiac anomalies and ventricular noncompaction produced by heterozygous Nkx2-5 homeodomain missense mutation. Circ. Cardiovasc. Genet. 2014, 7, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Ashraf, H.; Melanson, M.; Tanada, Y.; Nguyen, M.; Silberbach, M.; Wakimoto, H.; Benson, D.W.; Anderson, R.H.; Kasahara, H. Mouse model of human congenital heart disease: Progressive atrioventricular block induced by a heterozygous Nkx2-5 homeodomain missense mutation. Circ. Arrhythmia Electrophysiol. 2015, 8, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Stieber, J.; Herrmann, S.; Feil, S.; Löster, J.; Feil, R.; Biel, M.; Hofmann, F.; Ludwig, A. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc. Natl. Acad. Sci. USA 2003, 100, 15235–15240. [Google Scholar] [CrossRef] [Green Version]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Human Gene | Human CHD | Zebrafish Gene(s) | Zebrafish Phenotype | References |
|---|---|---|---|---|
| NR2F2 | AVSD, ASD, LVOTO, DORV, VSD | nr2f1a, nr2f2 | Nr2f1a: Atrial differentiation defects Nr2f1a; Nr2f2: Ventricular specification defects | [1,2,70,118,119,120] |
| NKX2.5 | ASD, VSD, ToF, Conduction defects | nkx2.5 | Defects in cardiomyocyte proliferation, differentiation, and maintenance, Conduction defects | [3,4,73,108,121,122,123,124] |
| TBX5 | ASD, VSD, Holt–Oram Syndrome | tbx5a, tbx5b | Looping defects, Bradycardia | [10,114,125,126] |
| SHOX2 | Atrial Fibrillation | shox2 | Bradycardia | [13,94,105,106] |
| HCN4 | Sick Sinus Syndrome | hcn4 | Bradycardia | [14,104,127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, K.E.; Waxman, J.S. Atrial and Sinoatrial Node Development in the Zebrafish Heart. J. Cardiovasc. Dev. Dis. 2021, 8, 15. https://doi.org/10.3390/jcdd8020015
Martin KE, Waxman JS. Atrial and Sinoatrial Node Development in the Zebrafish Heart. Journal of Cardiovascular Development and Disease. 2021; 8(2):15. https://doi.org/10.3390/jcdd8020015
Chicago/Turabian StyleMartin, Kendall E., and Joshua S. Waxman. 2021. "Atrial and Sinoatrial Node Development in the Zebrafish Heart" Journal of Cardiovascular Development and Disease 8, no. 2: 15. https://doi.org/10.3390/jcdd8020015
APA StyleMartin, K. E., & Waxman, J. S. (2021). Atrial and Sinoatrial Node Development in the Zebrafish Heart. Journal of Cardiovascular Development and Disease, 8(2), 15. https://doi.org/10.3390/jcdd8020015