
The Influence of Treatment with PCSK9 Inhibitors and Variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) Genes on the Corresponding Inflammatory Markers in Patients with Very High Lipoprotein(a) Levels

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Blood Collection and Laboratory Investigations
2.3. Genotyping
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Single Nucleotide Polymorphism Frequencies in Selected Genes of Inflammatory Markers
3.3. Plasma Levels of Inflammatory Markers at Enrollment and after Treatment with PCSK9 Inhibitors
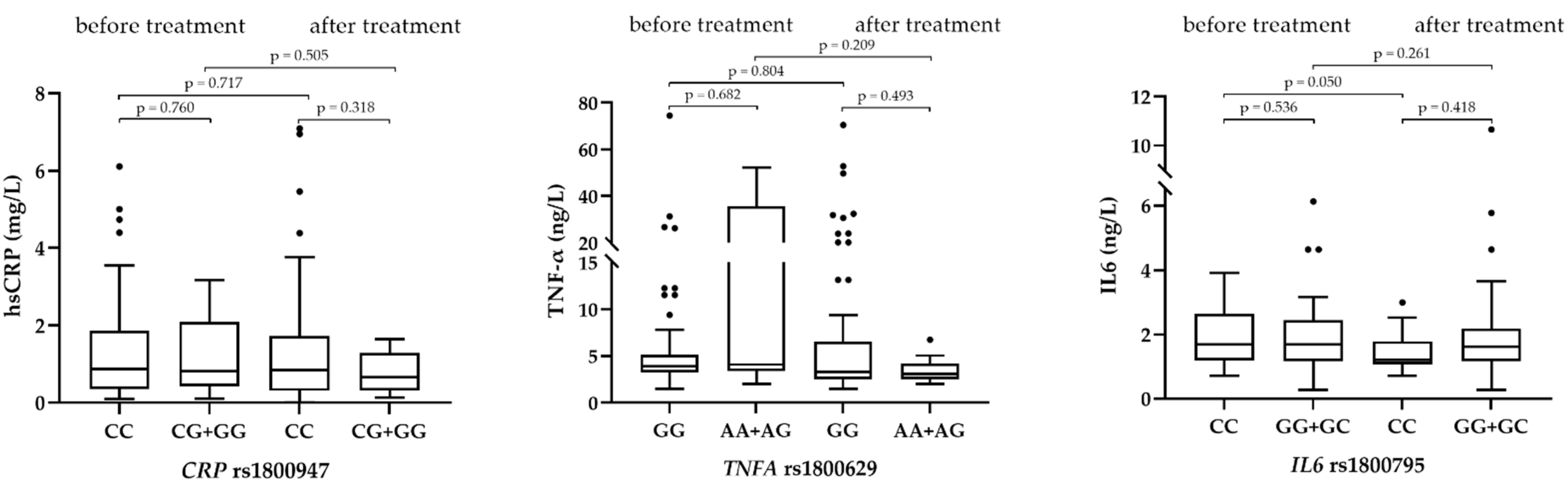
3.4. The Influence of Investigated Genotypes on Plasma Levels of Inflammatory Markers and Levels of Lipoprotein(a)
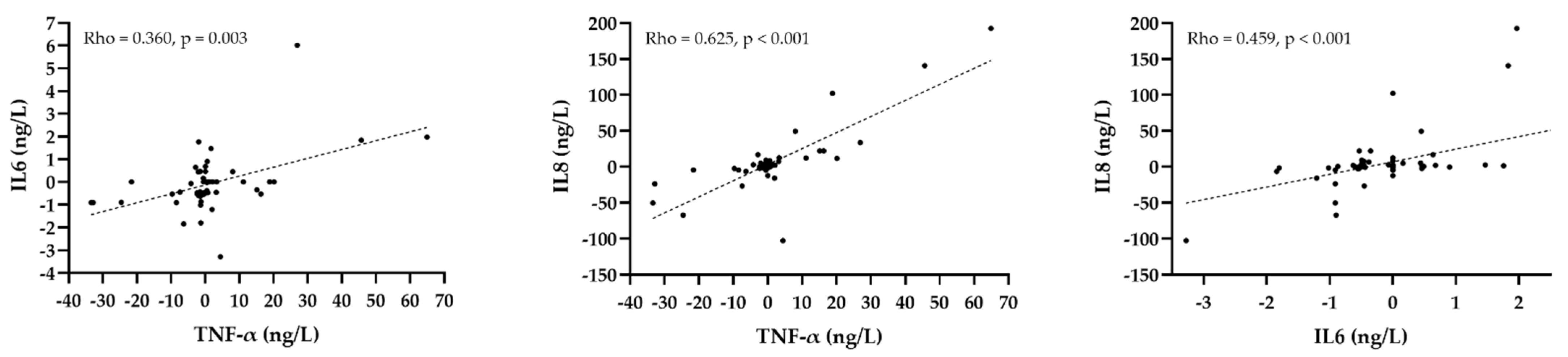
3.5. The Correlation of the Change of Inflammatory Markers with the Change of Lipid Parameters after Treatment with PCSK9 Inhibitors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ApoA1 | apolipoprotein A1 |
| ApoB | apolipoprotein B |
| CVD | cardiovascular disease |
| HDL-C | high-density lipoprotein cholesterol |
| HWE | Hardy-Weinberg equilibrium |
| hsCRP | high-sensitivity C-reactive protein |
| IL | interleukin |
| LDL-C | low-density lipoprotein cholestrerol |
| Lp(a) | lipoprotein(a) |
| MAF | minor allele frequency |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| SNP | single nucleotide polymorphism |
| TNF-α | tumor necrosis factor-α |
References
- Plutzky, J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. Am. J. Cardiol. 2001, 88, 10K–15K. [Google Scholar] [CrossRef]
- Fioranelli, M.; Bottaccioli, A.G.; Bottaccioli, F.; Bianchi, M.; Rovesti, M.; Roccia, M.G. Stress and inflammation in coronary artery disease: A review psychoneuroendocrineimmunology-based. Front. Immunol. 2018, 9, 2031. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Milani, R.V.; Verma, A.; O’Keefe, J.H. C-reactive protein and cardiovascular diseases—Is it ready for primetime? Am. J. Med. Sci. 2009, 338, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Grammer, T.B.; Marz, W.; Renner, W.; Bohm, B.O.; Hoffmann, M.M. C-reactive protein genotypes associated with circulating C-reactive protein but not with angiographic coronary artery disease: The LURIC study. Eur. Heart J. 2009, 30, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.I.; Fu, R.; Freeman, M.; Rogers, K.; Helfand, M. C-reactive protein as a risk factor for coronary heart disease: A systematic review and meta-analyses for the U.S. preventive services task force. Ann. Intern. Med. 2009, 151, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suleiman, M.; Khatib, R.; Agmon, Y.; Mahamid, R.; Boulos, M.; Kapeliovich, M.; Levy, Y.; Beyar, R.; Markiewicz, W.; Hammerman, H.; et al. Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction predictive role of C-reactive protein. J. Am. Coll. Cardiol. 2006, 47, 962–968. [Google Scholar] [CrossRef]
- Kathiresan, S.; Larson, M.G.; Vasan, R.S.; Guo, C.Y.; Gona, P.; Keaney, J.F., Jr.; Wilson, P.W.; Newton-Cheh, C.; Musone, S.L.; Camargo, A.L.; et al. Contribution of clinical correlates and 13 C-reactive protein gene polymorphisms to interindividual variability in serum C-reactive protein level. Circulation 2006, 113, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Worns, M.A.; Victor, A.; Galle, P.R.; Hohler, T. Genetic and environmental contributions to plasma C-reactive protein and interleukin-6 levels—A study in twins. Genes Immun. 2006, 7, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Pankow, J.S.; Folsom, A.R.; Cushman, M.; Borecki, I.B.; Hopkins, P.N.; Eckfeldt, J.H.; Tracy, R.P. Familial and genetic determinants of systemic markers of inflammation: The NHLBI family heart study. Atherosclerosis 2001, 154, 681–689. [Google Scholar] [CrossRef]
- MacGregor, A.J.; Gallimore, J.R.; Spector, T.D.; Pepys, M.B. Genetic effects on baseline values of C-reactive protein and serum amyloid a protein: A comparison of monozygotic and dizygotic twins. Clin. Chem. 2004, 50, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Díaz, Y.; Tovilla-Zárate, C.A.; Juárez-Rojop, I.; Baños-González, M.A.; Torres-Hernández, M.E.; López-Narváez, M.L.; Yañez-Rivera, T.G.; González-Castro, T.B. The role of gene variants of the inflammatory markers CRP and TNF-α in cardiovascular heart disease: Systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 11958–11984. [Google Scholar] [PubMed]
- Vaddi, K.; Nicolini, F.A.; Mehta, P.; Mehta, J.L. Increased secretion of tumor necrosis factor-alpha and interferon-gamma by mononuclear leukocytes in patients with ischemic heart disease. Relevance in superoxide anion generation. Circulation 1994, 90, 694–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barath, P.; Fishbein, M.C.; Cao, J.; Berenson, J.; Helfant, R.H.; Forrester, J.S. Detection and localization of tumor necrosis factor in human atheroma. Am. J. Cardiol. 1990, 65, 297–302. [Google Scholar] [CrossRef]
- Kroeger, K.M.; Carville, K.S.; Abraham, L.J. The -308 tumor necrosis factor alpha promoter polymorphism effects transcription. Mol. Immunol. 1997, 34, 391–399. [Google Scholar] [CrossRef]
- Lee, S.; Park, Y.; Zuidema, M.Y.; Hannink, M.; Zhang, C. Effects of interventions on oxidative stress and inflammation of cardiovascular diseases. World J. Cardiol. 2011, 3, 18–24. [Google Scholar] [CrossRef]
- Jenny, N.S.; Tracy, R.P.; Ogg, M.S.; Luong Le, A.; Kuller, L.H.; Arnold, A.M.; Sharrett, A.R.; Humphries, S.E. In the elderly, interleukin-6 plasma levels and the -174G>C polymorphism are associated with the development of cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2066–2071. [Google Scholar] [CrossRef] [Green Version]
- Riikola, A.; Sipila, K.; Kahonen, M.; Jula, A.; Nieminen, M.S.; Moilanen, L.; Kesaniemi, Y.A.; Lehtimaki, T.; Hulkkonen, J. Interleukin-6 promoter polymorphism and cardiovascular risk factors: The Health 2000 Survey. Atherosclerosis 2009, 207, 466–470. [Google Scholar] [CrossRef]
- Tan, J.; Hua, Q.; Li, J.; Fan, Z. Prognostic value of interleukin-6 during a 3-year follow-up in patients with acute ST-segment elevation myocardial infarction. Heart Vessel. 2009, 24, 329–334. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; Danesh, J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532 359 patients in the United States. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshar, M.; Pilote, L.; Dufresne, L.; Engert, J.C.; Thanassoulis, G. Lipoprotein(a) interactions with low-density lipoprotein cholesterol and other cardiovascular risk factors in premature acute coronary syndrome (ACS). J. Am. Heart Assoc. 2016, 5, e003012. [Google Scholar] [CrossRef] [Green Version]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- Desai, N.R.; Kohli, P.; Giugliano, R.P.; O’Donoghue, M.L.; Somaratne, R.; Zhou, J.; Hoffman, E.B.; Huang, F.; Rogers, W.J.; Wasserman, S.M.; et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy. Circulation 2013, 128, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Rehberger Likozar, A.; Blinc, A.; Trebušak Podkrajšek, K.; Šebeštjen, M. LPA genotypes and haplotypes are associated with lipoprotein(a) levels but not arterial wall properties in stable post-coronary event patients with very high lipoprotein(a) levels. J. Cardiovasc. Dev. Dis. 2021, 8, 181. [Google Scholar] [CrossRef]
- Baruch, A.; Mosesova, S.; Davis, J.D.; Budha, N.; Vilimovskij, A.; Kahn, R.; Peng, K.; Cowan, K.J.; Harris, L.P.; Gelzleichter, T.; et al. Effects of RG7652, a monoclonal antibody against PCSK9, on LDL-C, LDL-C subfractions, and inflammatory biomarkers in patients at high risk of or with established coronary heart disease (from the Phase 2 EQUATOR Study). Am. J. Cardiol. 2017, 119, 1576–1583. [Google Scholar] [CrossRef]
- Ascer, E.; Bertolami, M.C.; Venturinelli, M.L.; Buccheri, V.; Souza, J.; Nicolau, J.C.; Ramires, J.A.; Serrano, C.V., Jr. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis 2004, 177, 161–166. [Google Scholar] [CrossRef]
- Sahebkar, A.; Di Giosia, P.; Stamerra, C.A.; Grassi, D.; Pedone, C.; Ferretti, G.; Bacchetti, T.; Ferri, C.; Giorgini, P. Effect of monoclonal antibodies to PCSK9 on high-sensitivity C-reactive protein levels: A meta-analysis of 16 randomized controlled treatment arms. Br. J. Clin. Pharmacol. 2016, 81, 1175–1190. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Satny, M.; Hubacek, J.A.; Vrablik, M. Statins and inflammation. Curr. Atheroscler. Rep. 2021, 23, 80. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.; Danielson, E.; Fonseca, F.; Genest, J.; Gotto, A.J.; Kastelein, J.; Koenig, W.; Libby, P.; Lorenzatti, A.; MacFadyen, J.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, W. Low-grade inflammation modifies cardiovascular risk even at very low LDL-C levels: Are we aiming for a dual target concept? Circulation 2018, 138, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Libby, P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: Further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020, 41, 2153–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiekema, L.C.A.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; Fayad, Z.A. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2019, 40, 2775–2781. [Google Scholar] [CrossRef] [Green Version]
- Hoogeveen, R.M.; Opstal, T.S.J.; Kaiser, Y.; Stiekema, L.C.A.; Kroon, J.; Knol, R.J.J.; Bax, W.A.; Verberne, H.J.; Cornel, J.H.; Stroes, E.S.G. PCSK9 antibody alirocumab attenuates arterial wall inflammation without changes in circulating inflammatory markers. JACC Cardiovasc. Imaging 2019, 12, 2571–2573. [Google Scholar] [CrossRef]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory biomarkers interleukin-6 and C-reactive protein and outcomes in stable coronary heart disease: Experiences from the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) trial. J. Am. Heart Assoc. 2017, 6, e005077. [Google Scholar] [CrossRef] [Green Version]
- Vickers, M.A.; Green, F.R.; Terry, C.; Mayosi, B.M.; Julier, C.; Lathrop, M.; Ratcliffe, P.J.; Watkins, H.C.; Keavney, B. Genotype at a promoter polymorphism of the interleukin-6 gene is associated with baseline levels of plasma C-reactive protein. Cardiovasc. Res. 2002, 53, 1029–1034. [Google Scholar] [CrossRef] [Green Version]
- Araujo, F.; Pereira, A.C.; Mota, G.F.; Latorre Mdo, R.; Krieger, J.E.; Mansur, A.J. The influence of tumor necrosis factor -308 and C-reactive protein G1059C gene variants on serum concentration of C-reactive protein: Evidence for an age-dependent association. Clin. Chim. Acta 2004, 349, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Kolz, M.; Koenig, W.; Muller, M.; Andreani, M.; Greven, S.; Illig, T.; Khuseyinova, N.; Panagiotakos, D.; Pershagen, G.; Salomaa, V.; et al. DNA variants, plasma levels and variability of C-reactive protein in myocardial infarction survivors: Results from the AIRGENE study. Eur. Heart J. 2008, 29, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gomez, C.; Martin-Martinez, M.A.; Castaneda, S.; Sanchez-Alonso, F.; Uriarte-Ecenarro, M.; Gonzalez-Juanatey, C.; Romera-Baures, M.; Santos-Rey, J.; Pinto-Tasende, J.A.; Quesada-Masachs, E.; et al. Lipoprotein(a) concentrations in rheumatoid arthritis on biologic therapy: Results from the CARdiovascular in rheuMAtology study project. J. Clin. Lipidol. 2017, 11, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Thompson, L.; Giles, J.T.; Bathon, J.M.; Salmon, J.E.; Beaulieu, A.D.; Codding, C.E.; Carlson, T.H.; Delles, C.; Lee, J.S.; et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann. Rheum. Dis. 2015, 74, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Berthold, H.K.; Laudes, M.; Krone, W.; Gouni-Berthold, I. Association between the interleukin-6 promoter polymorphism -174G/C and serum lipoprotein(a) concentrations in humans. PLoS ONE 2011, 6, e24719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, N.; Schulte, D.M.; Turk, K.; Freitag-Wolf, S.; Hampe, J.; Zeuner, R.; Schroder, J.O.; Gouni-Berthold, I.; Berthold, H.K.; Krone, W.; et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J. Lipid Res. 2015, 56, 1034–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueland, T.; Kleveland, O.; Michelsen, A.E.; Wiseth, R.; Damas, J.K.; Holven, K.B.; Aukrust, P.; Gullestad, L.; Yndestad, A.; Halvorsen, B. Serum lipoprotein(a) is not modified by interleukin-6 receptor antagonism or associated with inflammation in non-ST-elevation myocardial infarction. Int. J. Cardiol. 2019, 274, 348–350. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Aday, A.W.; Rose, L.M.; Ridker, P.M. Residual inflammatory risk on treatment with PCSK9 inhibition and statin therapy. Circulation 2018, 138, 141–149. [Google Scholar] [CrossRef]
- Omer, W.; Naveed, A.K.; Khan, O.J.; Khan, D.A. Role of cytokine gene score in risk prediction of premature coronary artery disease. Genet. Test. Mol. Biomark. 2016, 20, 685–691. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Age at inclusion (years) | 52.6 (45.8–56.9) |
| Body mass index (kg/m2) | 28.6 ± 3.9 |
| Systolic blood pressure (mmHg) | 128 (120–135) |
| Diastolic blood pressure (mmHg) | 78 (70–82) |
| Heart rate (beat/min) | 61 (56–67) |
| Total cholesterol (mmol/L) | 4.24 ± 0.82 |
| HDL-C (mmol/L) | 1.20 ± 0.28 |
| Non-HDL-C (mmol/L) | 2.90 (2.40–3.68) |
| LDL-C (mmol/L) | 2.27 (1.69–2.66) |
| Triglycerides (mmol/L) | 1.43 (1.01–2.12) |
| Lp(a) (mg/L) | 1483 (1196–1785) |
| ApoA1 (g/L) | 1.28 (1.19–1.45) |
| ApoB (g/L) | 0.79 (0.67–0.99) |
| hsCRP (mg/L) | 0.87 (0.41–2.31) |
| TNF-α (ng/L) | 3.91 (3.13–5.14) |
| IL6 (ng/L) | 1.69 (1.20–2.18) |
| IL8 (ng/L) | 13.5 (10.6–17.8) |
| SNP | Genotype | Number (%) | p Value HWE | MAF |
|---|---|---|---|---|
| CRP rs1800947 | CC | 58 (84.1) | 1.000 | 0.087 |
| CG | 10 (14.5) | |||
| GG | 1 (1.4) | |||
| TNFA rs1800629 | GG | 58 (84.1) | 1.000 | 0.087 |
| GA | 10 (14.5) | |||
| AA | 1 (1.4) | |||
| IL6 rs1800795 | CC | 16 (23.2) | 0.988 | 0.478 |
| GC | 34 (49.3) | |||
| GG | 19 (27.5) |
| Inflammatory Marker | Group | Baseline | After 6 Months | p Value |
|---|---|---|---|---|
| hsCRP (mg/L) | Control | 0.80 (0.39–2.23) | 0.74 (0.35–1.47) | 0.713 |
| Treatment | 0.85 (0.36–1.81) | 0.79 (0.31–1.57) | 0.989 | |
| TNF-α (ng/L) | Control | 3.74 (2.99–5.02) | 3.91 (3.05–5.25) | 0.396 |
| Treatment | 3.91 (3.36–5.36) | 3.30 (2.53–6.04) | 0.402 | |
| IL6 (ng/L) | Control | 1.69 (1.19–2.18) | 1.69 (1.19–2.75) | 0.614 |
| Treatment | 1.69 (1.20–2.53) | 1.36 (1.16–2.18) | 0.066 | |
| IL8 (ng/L) | Control | 11.8 (10.8–16.1) | 11.5 (10.8–16.3) | 0.819 |
| Treatment | 13.5 (10.7–17.7) | 15.5 (10.8–24.1) | 0.250 |
| SNP | Genotype | Median (25–75%) | p Value |
|---|---|---|---|
| CRP rs1800947 | CC | 0.93 (0.41–2.10) mg/L | 0.664 |
| CG+GG | 0.81 (0.40–2.38) mg/L | ||
| TNFA rs1800629 | GG | 3.91 (3.05–5.14) ng/L | 0.555 |
| GA+AA | 3.83 (3.28–35.27) ng/L | ||
| IL6 rs1800795 | CC | 1.69 (1.20–2.07) ng/L | 0.888 |
| CG+GG | 1.69 (1.18–2.36) ng/L |
| SNP | Genotype | Median (25–75%) | p Value |
|---|---|---|---|
| CRP rs1800947 | GG+GC | 1559 (974–1782) | 0.782 |
| CC | 1463 (1196–1837) | ||
| TNFA rs1800629 | AA+AG | 1607 (1219–2105) | 0.376 |
| GG | 1462 (1171–1741) | ||
| IL6 rs1800795 | GG+CG | 1461 (1011–1785) | 0.316 |
| CC | 1580 (1295–1871) |
| Parameter | hsCRP | TNF-α | IL6 | IL8 | ||||
|---|---|---|---|---|---|---|---|---|
| Rho | p Value | Rho | p Value | Rho | p Value | Rho | p Value | |
| Total cholesterol | −0.022 | 0.863 | −0.081 | 0.519 | 0.061 | 0.629 | 0.056 | 0.656 |
| HDL-C | −0.138 | 0.272 | −0.037 | 0.769 | 0.091 | 0.471 | −0.015 | 0.904 |
| LDL-C | 0.044 | 0.726 | −0.009 | 0.946 | 0.103 | 0.415 | 0.087 | 0.489 |
| Triglycerides | −0.019 | 0.881 | −0.097 | 0.443 | −0.077 | 0.543 | −0.077 | 0.543 |
| Lp(a) | 0.104 | 0.419 | −0.168 | 0.188 | −0.227 | 0.073 | −0.076 | 0.552 |
| ApoB | 0.009 | 0.945 | −0.097 | 0.453 | 0.031 | 0.808 | 0.064 | 0.623 |
| ApoA1 | −0.116 | 0.371 | −0.166 | 0.198 | −0.077 | 0.549 | 0.098 | 0.449 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levstek, T.; Podkrajšek, N.; Rehberger Likozar, A.; Šebeštjen, M.; Trebušak Podkrajšek, K. The Influence of Treatment with PCSK9 Inhibitors and Variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) Genes on the Corresponding Inflammatory Markers in Patients with Very High Lipoprotein(a) Levels. J. Cardiovasc. Dev. Dis. 2022, 9, 127. https://doi.org/10.3390/jcdd9050127
Levstek T, Podkrajšek N, Rehberger Likozar A, Šebeštjen M, Trebušak Podkrajšek K. The Influence of Treatment with PCSK9 Inhibitors and Variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) Genes on the Corresponding Inflammatory Markers in Patients with Very High Lipoprotein(a) Levels. Journal of Cardiovascular Development and Disease. 2022; 9(5):127. https://doi.org/10.3390/jcdd9050127
Chicago/Turabian StyleLevstek, Tina, Nik Podkrajšek, Andreja Rehberger Likozar, Miran Šebeštjen, and Katarina Trebušak Podkrajšek. 2022. "The Influence of Treatment with PCSK9 Inhibitors and Variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) Genes on the Corresponding Inflammatory Markers in Patients with Very High Lipoprotein(a) Levels" Journal of Cardiovascular Development and Disease 9, no. 5: 127. https://doi.org/10.3390/jcdd9050127
APA StyleLevstek, T., Podkrajšek, N., Rehberger Likozar, A., Šebeštjen, M., & Trebušak Podkrajšek, K. (2022). The Influence of Treatment with PCSK9 Inhibitors and Variants in the CRP (rs1800947), TNFA (rs1800629), and IL6 (rs1800795) Genes on the Corresponding Inflammatory Markers in Patients with Very High Lipoprotein(a) Levels. Journal of Cardiovascular Development and Disease, 9(5), 127. https://doi.org/10.3390/jcdd9050127