
A Comprehensive Assessment of Ultraviolet-Radiation-Induced Mutations in Flammulina filiformis Using Whole-Genome Resequencing
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Oidium Collection
2.2. Mutation Experiment
2.3. Whole-Genome Resequencing
2.4. Base Change Calling and Distributions of Single-Base Mutations on Contigs
2.5. Effects of Immediately Adjacent Bases on Mutation Frequency and Tandem Mutation
2.6. Detection of DNA Fragment Deletion
2.7. Functional Annotation of Mutated Genes
3. Results
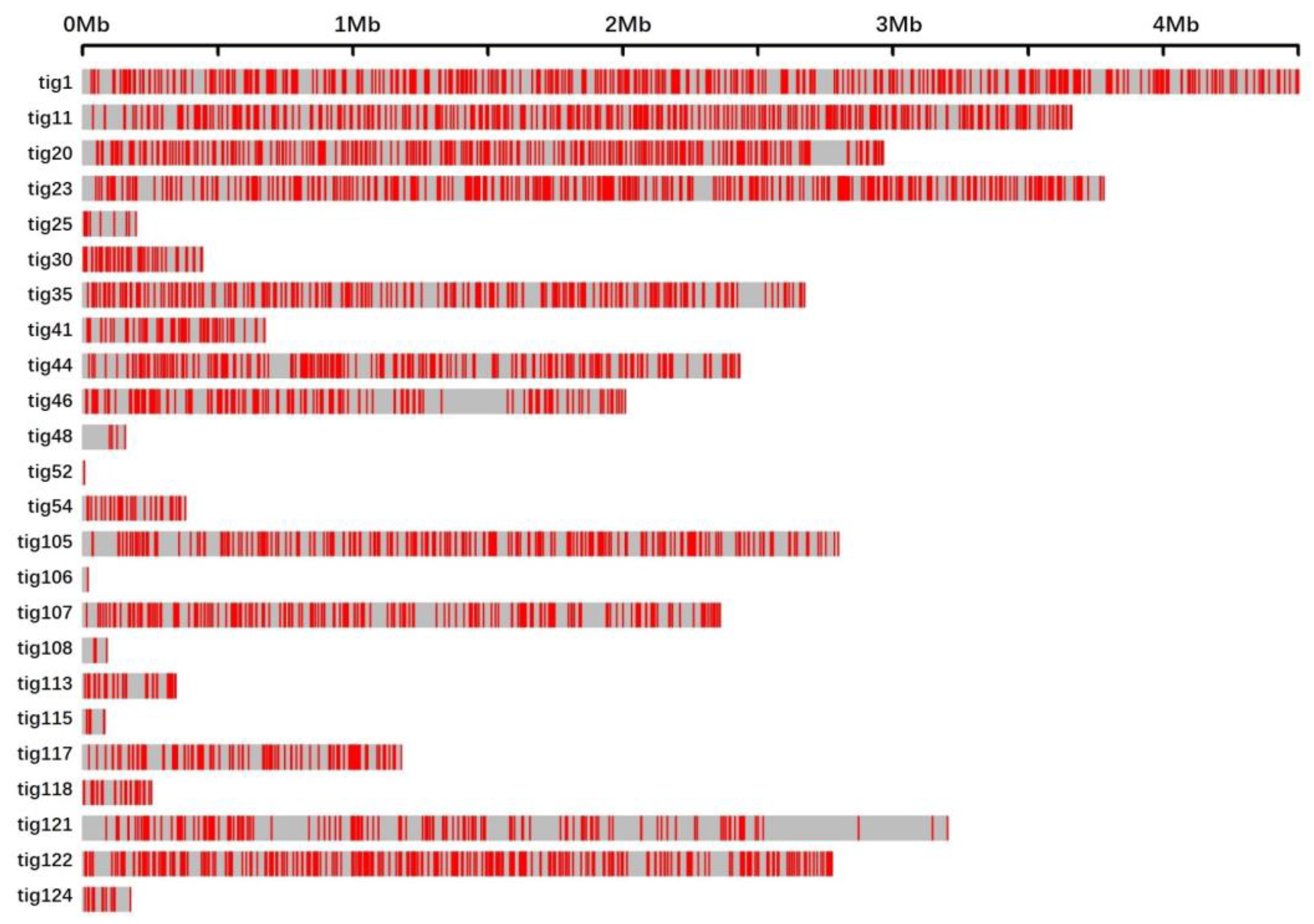
3.1. Single-Base Substitutions and Their Distributions on Contigs
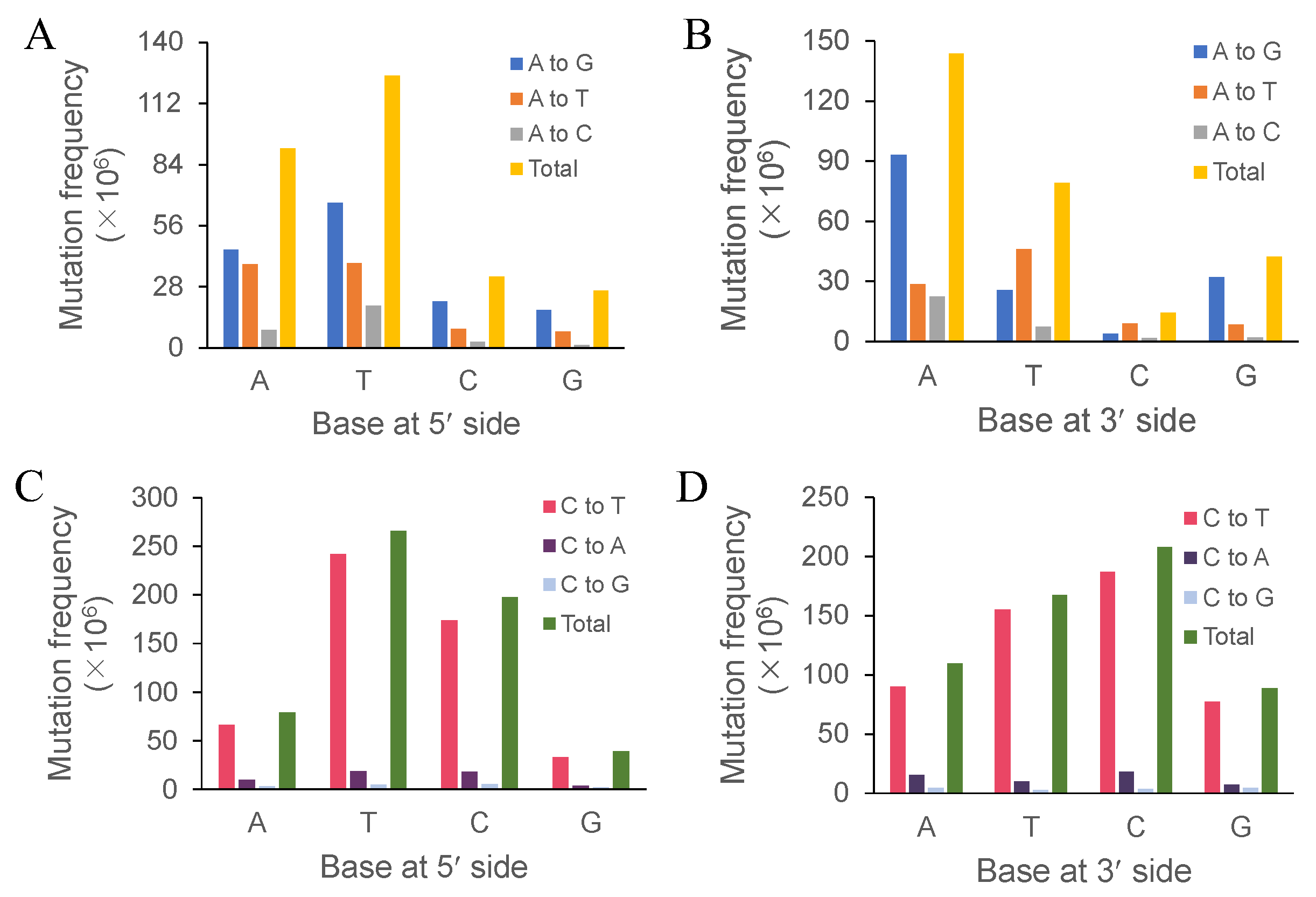
3.2. Effects of Immediately Adjacent Bases on Mutation Frequency
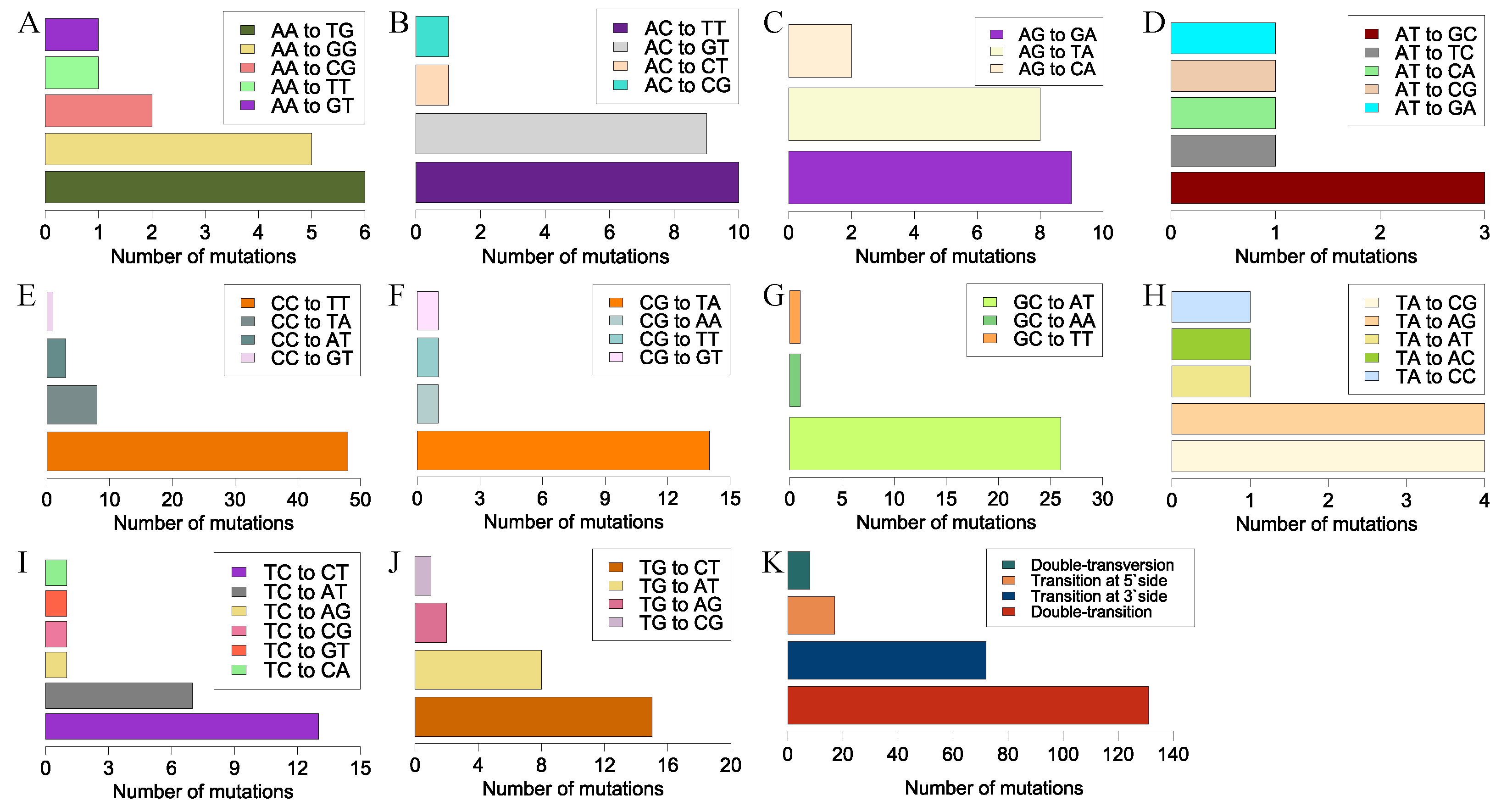
3.3. Tandem Mutations
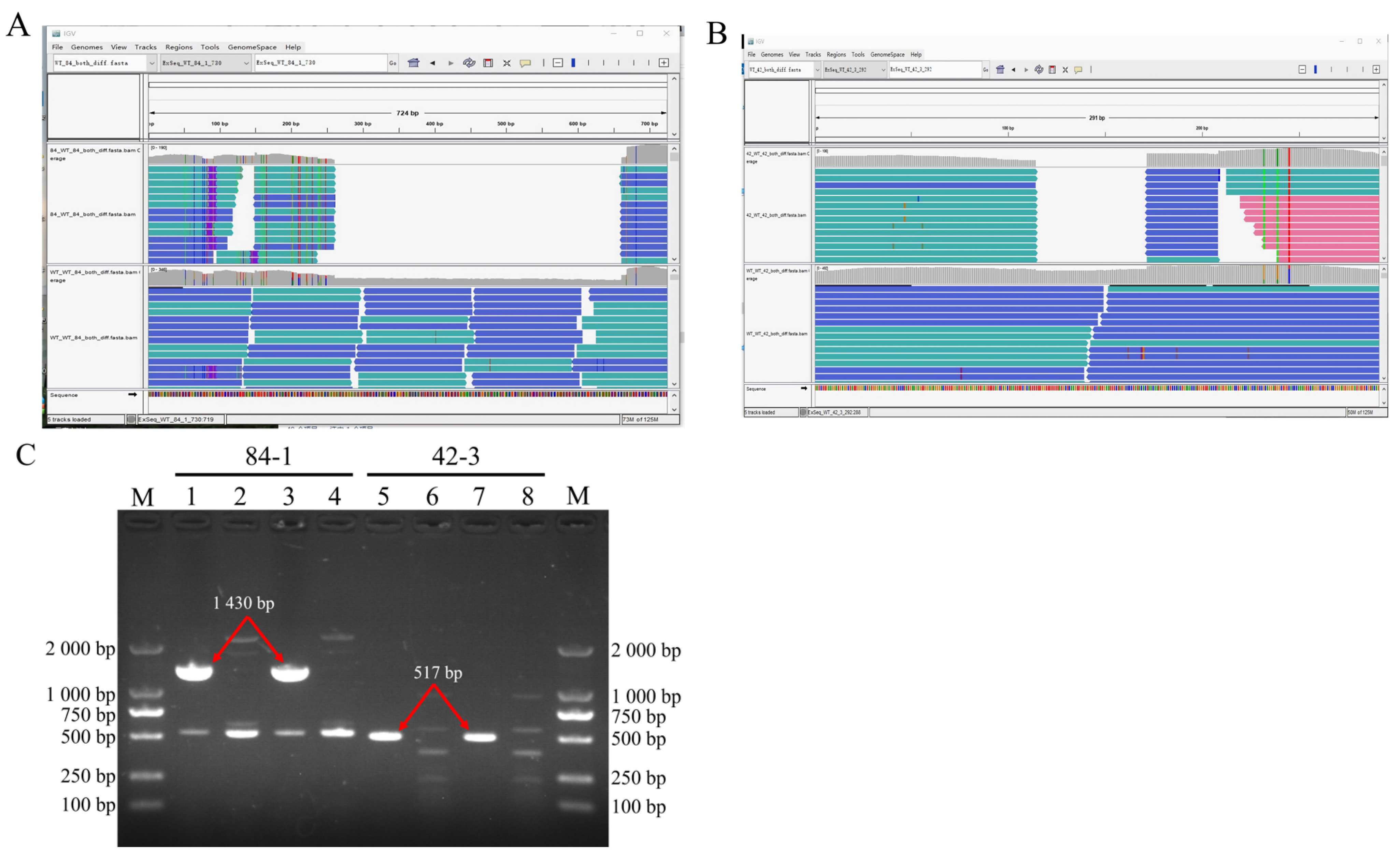
3.4. DNA Fragment Deletions
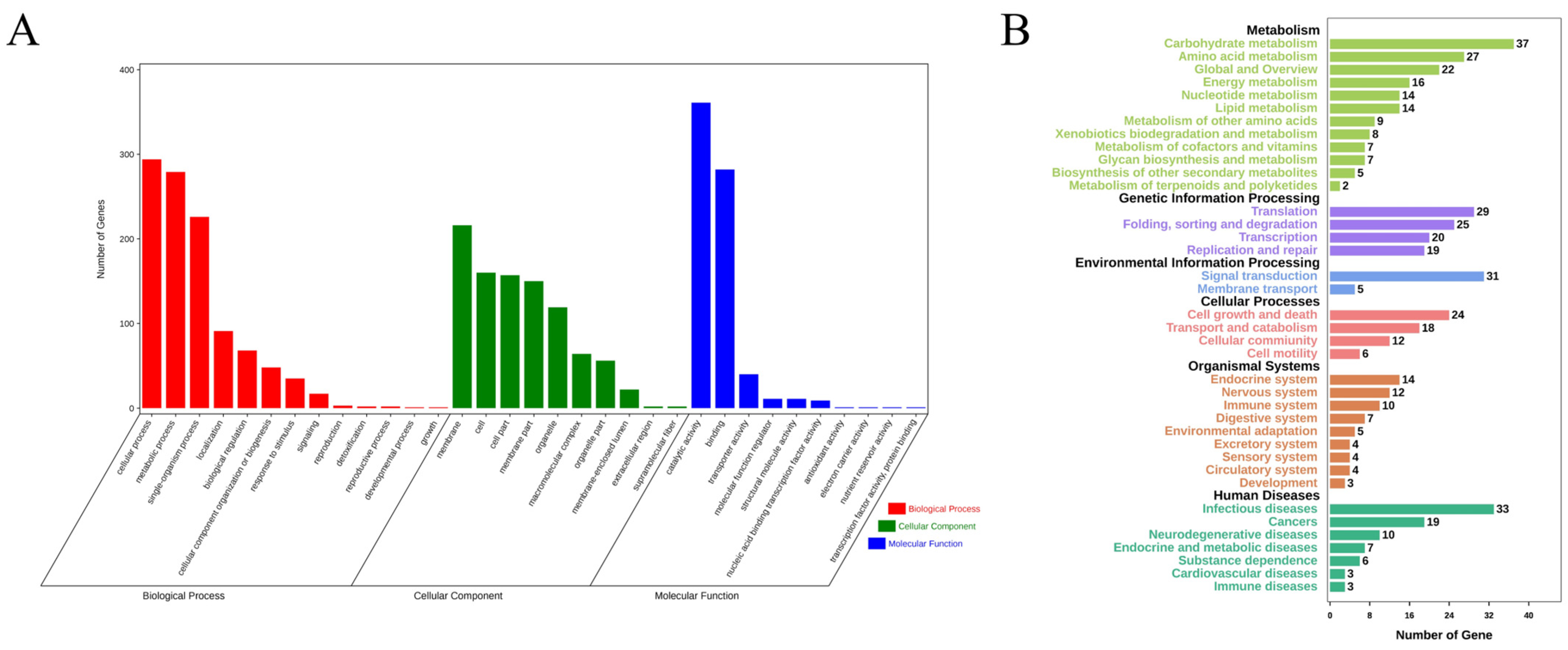
3.5. Annotation of Genes with Non-Synonymous Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wakeley, J. Substitution-rate variation among sites and the estimation of transition bias. Mol. Biol. Evol. 1994, 11, 436–442. [Google Scholar]
- Yang, Z.; Yoder, A.D. Estimation of the transition/transversion rate bias and species sampling. J. Mol. Evol. 1999, 48, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. 2005, 571, 19–31. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Farlow, A.; Long, H.A.; Arnoux, S.; Sung, W.; Doak, T.G.; Nordborg, M.; Lynch, M. The Spontaneous Mutation Rate in the Fission Yeast Schizosaccharomyces pombe. Genetics 2015, 201, 737–744. [Google Scholar] [CrossRef]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef]
- Hartman, P.S.; Hlavacek, A.; Wilde, H.; Lewicki, D.; Schubert, W.; Kern, R.G.; Kazarians, G.A.; Benton, E.V.; Benton, E.R.; Nelson, G.A. A comparison of mutations induced by accelerated iron particles versus those induced by low earth orbit space radiation in the FEM-3 gene of Caenorhabditis elegans. Mutat. Res. 2001, 474, 47–55. [Google Scholar] [CrossRef]
- Sun, M.; Mondal, K.; Patel, V.; Horner, V.L.; Long, A.B.; Cutler, D.J.; Caspary, T.; Zwick, M.E. Multiplex chromosomal exome sequencing accelerates identification of ENU-induced mutations in the mouse. G3 2012, 2, 143–150. [Google Scholar] [CrossRef]
- Thacker, J.; Stretch, A.; Stephens, M.A. Mutation and inactivation of cultured mammalian cells exposed to beams of accelerated heavy ions: II. Chinese hamster V79 cells. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1979, 36, 137–148. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Ikehata, H.; Ono, T. The mechanisms of UV mutagenesis. J Radiat. Res. 2011, 52, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Grand, A.; Douki, T. Solar UV radiation-induced DNA bipyrimidine photoproducts: Formation and mechanistic insights. In Photoinduced Phenomena in Nucleic Acids II; Topics in Current Chemistry; Barbatii, M., Borin, A., Ullrich, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 356, pp. 249–275. [Google Scholar]
- Gallagher, R.P.; Lee, T.K.; Bajdik, C.D.; Borugian, M. Ultraviolet radiation. Chron. Dis. Inj. Can. 2010, 29 (Suppl. 1), 51–68. [Google Scholar] [CrossRef]
- Kappes, U.P.; Luo, D.; Potter, M.; Schulmeister, K.; Runger, T.M. Short-and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J. Investig. Dermatol. 2006, 126, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Coulondre, C.; Miller, J.H. Genetic studies of the lac repressor: IV. Mutagenic specificity in the lacI gene of Escherichia coli. J. Mol. Biol. 1977, 117, 577–606. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.H. Mutagenic specificity of ultraviolet light. J. Mol. Biol. 1985, 182, 45–65. [Google Scholar] [CrossRef]
- Brash, D.E. UV signature mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Formation and processing of UV photoproducts: Effects of DNA sequence and chromatin environment. Photochem. Photobiol. 1997, 65, 270–283. [Google Scholar] [CrossRef]
- Darr, D.; Fridovich, I. Free radicals in cutaneous biology. J. Investig. Dermatol. 1994, 102, 671–675. [Google Scholar] [CrossRef]
- Stary, A.; Sarasin, A. Ultraviolet A-and singlet oxygen-induced mutation spectra. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2000; Volume 319, pp. 153–165. [Google Scholar]
- Livneh, Z.; Cohen-Fix, O.; Skaliter, R.; Elizur, T. Replication of damaged DNA and the molecular mechanism of ultraviolet light mutagenesis. Crit. Rev. Biochem. Mol. 1993, 28, 465–513. [Google Scholar] [CrossRef]
- Qu, L.J.; Foote, T.N.; Roberts, M.A.; Money, T.A.; Aragon-Alcaide, L.; Snape, J.W.; Moore, G. A simple PCR-based method for scoring the ph1b deletion in wheat. Theor. Appl. Genet. 1998, 96, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.M.; Smerdon, M.J. UV induced (6-4) photoproducts are distributed differently than cyclobutane dimers in nucleosomes. Photochem. Photobiol. 1990, 51, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Bourre, F.; Renault, G.; Seawell, P.C.; Sarasin, A. Distribution of ultraviolet-induced lesions in simian virus 40 DNA. Biochimie 1985, 67, 293–299. [Google Scholar] [CrossRef]
- Lippke, J.A.; Gordon, L.K.; Brash, D.E.; Haseltine, W.A. Distribution of UV light-induced damage in a defined sequence of human DNA: Detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3388–3392. [Google Scholar] [CrossRef] [PubMed]
- Keohavong, P.; Liu, V.F.; Thilly, W.G. Analysis of point mutations induced by ultraviolet light in human cells. Mutat. Res. 1991, 249, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Rozek, D.; Pfeifer, G.P. The distribution of UV photoproducts along the human p53 gene and its relation to mutations in skin cancer. Oncogene 1993, 8, 2051–2057. [Google Scholar]
- Besaratinia, A.; Synold, T.W.; Xi, B.; Pfeifer, G.P. G-to-T transversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet a radiation in mammalian cells. Biochemistry 2004, 43, 8169–8177. [Google Scholar] [CrossRef]
- Hauser, J.; Seidman, M.M.; Sidur, K.; Dixon, K. Sequence specificity of point mutations induced during passage of a UV-irradiated shuttle vector plasmid in monkey cells. Mol. Cell. Biol. 1986, 6, 277–285. [Google Scholar]
- Shibai, A.; Takahashi, Y.; Ishizawa, Y.; Motooka, D.; Nakamura, S.; Ying, B.W.; Tsuru, S. Mutation accumulation under UV radiation in Escherichia coli. Sci. Rep. 2017, 7, 14531. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef]
- Kemp, R.F.O. Production of oidia by dikaryons of Flammulina velutipes. Trans. Br. Mycol. Soc. 1980, 74, 557–560. [Google Scholar] [CrossRef]
- Wang, W.; Liu, F.; Jiang, Y.; Wu, G.; Guo, L.; Chen, R.; Chen, B.; Lu, Y.; Dai, Y.; Xie, B. The multigene family of fungal laccases and their expression in the white rot basidiomycete Flammulina velutipes. Gene 2015, 563, 142–149. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis Toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Bergland, A.O.; Gonzalez, J.; Petrov, D.A. Empirical validation of pooled whole genome population re-sequencing in Drosophila melanogaster. PLoS ONE 2012, 7, e41901. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.Z.; Wang, M.; Zhang, P.; Liu, F.; Yan, J.J.; Xie, B.G. Identification of Pleurotus eryngii Varieties by MNP Makers Based on Next-Generation Sequencing. Acta Edulis Fungi 2023, 30, 1–9. [Google Scholar]
- Lomsadze, A.; Ter-Hovhannisyan, V.; Chernoff, Y.O.; Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33, 6494–6506. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Protić-Sabljić, M.; Tuteja, N.; Munson, P.J.; Hauser, J.; Kraemer, K.H.; Dixon, K. UV light-induced cyclobutane pyrimidine dimers are mutagenic in mammalian cells. Mol. Cell. Biol. 1986, 6, 3349–3356. [Google Scholar]
- Schaaper, R.M.; Dunn, R.L.; Glickman, B.W. Mechanisms of ultraviolet-induced mutation: Mutational spectra in the Escherichia coli lacI gene for a wild-type and an excision-repair-deficient strain. J. Mol. Biol. 1987, 198, 187–202. [Google Scholar] [CrossRef]
- Fowler, R.G.; McGinty, L.; Mortelmans, K.E. Mutational specificity of ultraviolet light in Escherichia coli with and without the R plasmid pKM101. Genetics 1981, 99, 25–40. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Allison, J.P.; Nairn, R.S. Immunoprecipitation of pyrimidine (6–4) pyrimidone photoproducts and cyclobutane pyrimidine dimers in UV-irradiated DNA. Radiat. Res. 1990, 123, 299–303. [Google Scholar] [CrossRef]
- Yoon, J.H.; Lee, C.S.; O’Connor, T.R.; Yasui, A.; Pfeifer, G.P. The DNA damage spectrum produced by simulated sunlight. J. Mol. Biol. 2000, 299, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Drobetsky, E.A.; Grosovsky, A.J.; Glickman, B.W. The specificity of UV-induced mutations at an endogenous locus in mammalian cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9103–9107. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Iwai, S.; Kasai, H. The (6–4) photoproduct of thymine-thymine induces targeted substitution mutations in mammalian cells. Nucleic Acids Res. 1998, 26, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Gentil, A.; Le Page, F.; Margot, A.; Lawrence, C.W.; Borden, A.; Sarasin, A. Mutagenicity of a unique thymine-thymine dimer or thymine-thymine pyrimidine pyrimidone (6–4) photoproduct in mammalian cells. Nucleic Acids Res. 1996, 24, 1837–1840. [Google Scholar] [CrossRef]
- Koch, R.E. The influence of neighboring base pairs upon base-pair substitution mutation rates. Proc. Natl. Acad. Sci. USA 1971, 68, 773–776. [Google Scholar] [CrossRef]
- Brash, D.E.; Seetharam, S.; Kraemer, K.H.; Seidman, M.M.; Bredberg, A. Photoproduct frequency is not the major determinant of UV base substitution hot spots or cold spots in human cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3782–3786. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Drouin, R.; Riggs, A.D.; Holmquist, G.P. In vivo mapping of a DNA adduct at nucleotide resolution: Detection of pyrimidine (6–4) pyrimidone photoproducts by ligation-mediated polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1991, 88, 1374–1378. [Google Scholar] [CrossRef]
- Lee, D.H.; Pfeifer, G.P. Deamination of 5-Methylcytosines within Cyclobutane Pyrimidine Dimers Is an Important Component of UVB Mutagenesis. J. Biol. Chem. 2003, 278, 10314–10321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Q.; Li, J.; Zeng, C.; Hu, S.; Yu, J. The study of neighboring nucleotide composition and transition/transversion bias. Sci. China Ser. C Life Sci. 2006, 49, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Morton, B.R. The rolf of context-dependent mutation in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 2003, 56, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Radman, M.; Wagner, R. Mismatch repair in Escherichia coli. Ann. Rev. Genet. 1986, 20, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.W.; Chou, S.H.; Reid, B.R. Base pairing geometry in GA mismatches depends entirely on the neighboring sequence. J. Mol. Biol. 1992, 228, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.W.; Libertini, L.J.; Suquet, C.; Small, E.W.; Smerdon, M.J. Unfolding of Nucleosome Cores Dramatically Changes the Distribution of Ultraviolet Photoproducts in DNA. Biochemistry 1993, 32, 10527–10531. [Google Scholar] [CrossRef]
- Brown, A.J.; Mao, P.; Smerdon, M.J.; Wyrick, J.J.; Roberts, S.A. Nucleosome positions establish an extended mutation signature in melanoma. PLoS Genet. 2018, 14, e1007823. [Google Scholar] [CrossRef]
- Drouin, R.; Therrien, J.P. UVB-induced Cyclobutane Pyrimidine Dimer Frequency Correlates with Skin Cancer Mutational Hotspots in p53. Photochem. Photobiol. 1997, 66, 719–726. [Google Scholar] [CrossRef]
- Nakazawa, H.; English, D.; Randell, P.L.; Nakazawa, K.; Martel, N.; Armstrong, B.K.; Yamasaki, H. UV and skin cancer: Specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc. Natl. Acad. Sci. USA 1994, 91, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar] [CrossRef]
- Bredberg, A.; Kraemer, K.H.; Seidman, M.M. Restricted ultraviolet mutational spectrum in a shuttle vector propagated in xeroderma pigmentosum cells. Proc. Natl. Acad. Sci. USA 1986, 83, 8273–8277. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Dammann, R.; Pfeifer, G.P. Sequence and Time-Dependent Deamination of Cytosine Bases in UVB-Induced Cyclobutane Pyrimidine Dimers In Vivo. J. Mol. Biol. 1998, 284, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Scherer, S.W.; Lee, C.; Birney, E.; Altshuler, D.M.; Eichler, E.E.; Carter, N.P.; Hurles, E.M.; Feuk, L. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 2007, 39 (Suppl. 7), S7–S15. [Google Scholar] [CrossRef]
- Corrigan, M.W.; Kerwin-Iosue, C.L.; Kuczmarski, A.S.; Amin, K.B.; Wykoff, D.D. The Fate of Linear DNA in Saccharomyces cerevisiae and Candida glabrata: The Role of Homologous and Non-Homologous End Joining. PLoS ONE 2013, 8, e69628. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quality Index | Average | Minimum |
|---|---|---|
| Seq_depth (✕) | 65.39 | 35.93 |
| Q20 (%) | 96.07 | 95.02 |
| Q30 (%) | 90.73 | 88.71 |
| GC content (%) | 48.31 | 42.62 |
| Substitution Types | No. of Mutation Sites | Ratio |
|---|---|---|
| G:C→A:T | 2239 | 60.40% |
| A:T→G:C | 651 | 17.56% |
| A:T→T:A | 394 | 10.63% |
| G:C→T:A | 221 | 5.96% |
| A:T→C:G | 135 | 3.64% |
| G:C→C:G | 67 | 1.81% |
| Total | 3707 | 100% |
| Contig | Mutated Sites per Contig | Contig Length (bp) | Average Length between Mutated Sites |
|---|---|---|---|
| tig1 | 473 | 4,475,816 | 9463 |
| tig11 | 427 | 3,630,557 | 8502 |
| tig14 | 0 | 48,161 | - |
| tig15 | 0 | 62,023 | - |
| tig18 | 0 | 36,591 | - |
| tig20 | 316 | 3,033,355 | 9599 |
| tig23 | 425 | 3,725,332 | 8765 |
| tig25 | 14 | 222,816 | 15,915 |
| tig30 | 55 | 443,364 | 8061 |
| tig35 | 262 | 2,648,354 | 10,108 |
| tig41 | 0 | 678,856 | - |
| tig44 | 237 | 2,393,417 | 10,099 |
| tig46 | 175 | 2,040,433 | 11,660 |
| tig48 | 5 | 155,795 | 31,159 |
| tig52 | 1 | 48,353 | 48,353 |
| tig54 | 38 | 386,978 | 10,184 |
| tig56 | 0 | 119,340 | - |
| tig105 | 269 | 2,754,843 | 10,241 |
| tig106 | 1 | 37,686 | 37,686 |
| tig107 | 220 | 2,350,230 | 10,683 |
| tig108 | 3 | 147,222 | 49,074 |
| tig109 | 0 | 23,566 | - |
| tig110 | 0 | 137,946 | - |
| tig111 | 0 | 78,587 | - |
| tig112 | 0 | 184,367 | - |
| tig113 | 40 | 370,450 | 9261 |
| tig115 | 6 | 100,769 | 16,795 |
| tig117 | 128 | 1,207,482 | 9433 |
| tig118 | 36 | 276,954 | 7693 |
| tig121 | 177 | 3,240,382 | 18,307 |
| tig122 | 319 | 2,748,975 | 8617 |
| tig124 | 16 | 222,876 | 13,930 |
| Bases at 3′ | Muation Types | Total | Chi-square Test | |
|---|---|---|---|---|
| A→G | A→T | |||
| A+G | 523 | 153 | 676 | p = 0.000 |
| T+C | 128 | 241 | 369 | |
| Total | 651 | 394 | 1045 | |
| Mutation Region | Mutation Frequency | Proportion |
|---|---|---|
| Intergenic region | 627 | 16.91% |
| Intron | 633 | 17.08% |
| Exon (non-synonymous) | 1458 | 39.33% |
| Exon (synonymous) | 707 | 19.07% |
| Core promoter | 282 | 7.61% |
| KEGG B Class | Pathway | Pathway ID | Gene Number |
|---|---|---|---|
| Carbohydrate metabolism | Amino sugar and nucleotide sugar metabolism | ko00520 | 10 |
| Starch and sucrose metabolism | ko00500 | 7 | |
| Inositol phosphate metabolism | ko00562 | 6 | |
| Amino acid metabolism | Valine, leucine and isoleucine degradation | ko00280 | 6 |
| Glycine, serine and threonine metablism | ko00260 | 4 | |
| Tyrosine metabolism | ko00350 | 4 | |
| Alamine, aspartate and glutamate metabolism | ko00250 | 4 | |
| Translation | Ribosome biogenesis in eukaryotes | ko03008 | 7 |
| mRNA surveillance pathway | ko03015 | 5 | |
| Folding, sorting and degradation | Protein processing in endoplasmic reticulum | ko04141 | 8 |
| Ubiquitin mediated proteolysis | ko04120 | 7 | |
| RNA degradation | ko03018 | 7 | |
| Transcription | Spliceosome | ko03040 | 13 |
| Basal transcription factors | ko03022 | 5 | |
| Signal transduction | Ras signaling pathway | ko04014 | 5 |
| MAPK signaling pathway | ko04010 | 5 | |
| Phosphatidylinositol signaling syStem | ko04070 | 5 | |
| PI3K-Akt signaling pathway | ko04151 | 5 | |
| Wnt signaling pathway | ko04310 | 5 | |
| Cell growth and death | Cell cycle - yeast | ko04111 | 15 |
| Meiosis - yeast | ko04113 | 11 | |
| Cell cycle | ko04110 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Han, X.; Tong, Z.; Deng, Y.; Xie, L.; Liu, S.; Xie, B.; Zhang, W. A Comprehensive Assessment of Ultraviolet-Radiation-Induced Mutations in Flammulina filiformis Using Whole-Genome Resequencing. J. Fungi 2024, 10, 228. https://doi.org/10.3390/jof10030228
Huang Q, Han X, Tong Z, Deng Y, Xie L, Liu S, Xie B, Zhang W. A Comprehensive Assessment of Ultraviolet-Radiation-Induced Mutations in Flammulina filiformis Using Whole-Genome Resequencing. Journal of Fungi. 2024; 10(3):228. https://doi.org/10.3390/jof10030228
Chicago/Turabian StyleHuang, Qianhui, Xing Han, Zongjun Tong, Youjin Deng, Luyu Xie, Shengrong Liu, Baogui Xie, and Weirui Zhang. 2024. "A Comprehensive Assessment of Ultraviolet-Radiation-Induced Mutations in Flammulina filiformis Using Whole-Genome Resequencing" Journal of Fungi 10, no. 3: 228. https://doi.org/10.3390/jof10030228
APA StyleHuang, Q., Han, X., Tong, Z., Deng, Y., Xie, L., Liu, S., Xie, B., & Zhang, W. (2024). A Comprehensive Assessment of Ultraviolet-Radiation-Induced Mutations in Flammulina filiformis Using Whole-Genome Resequencing. Journal of Fungi, 10(3), 228. https://doi.org/10.3390/jof10030228