Biological Roles of Protein-Coding Tandem Repeats in the Yeast Candida Albicans


Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Tandem Repeats in the C. albicans Genome
2.2. Analysis of Tandem Repeats in Other Genomes
2.3. Determination of Allele Distributions for Six TR Regions
3. Results and Discussion
3.1. Almost Half of the ORFs in the C. albicans Genome Are TR-ORFs
3.2. TR-ORFs Considerably Increase the Genome-Wide Frequency of Mutations in C. albicans Exons
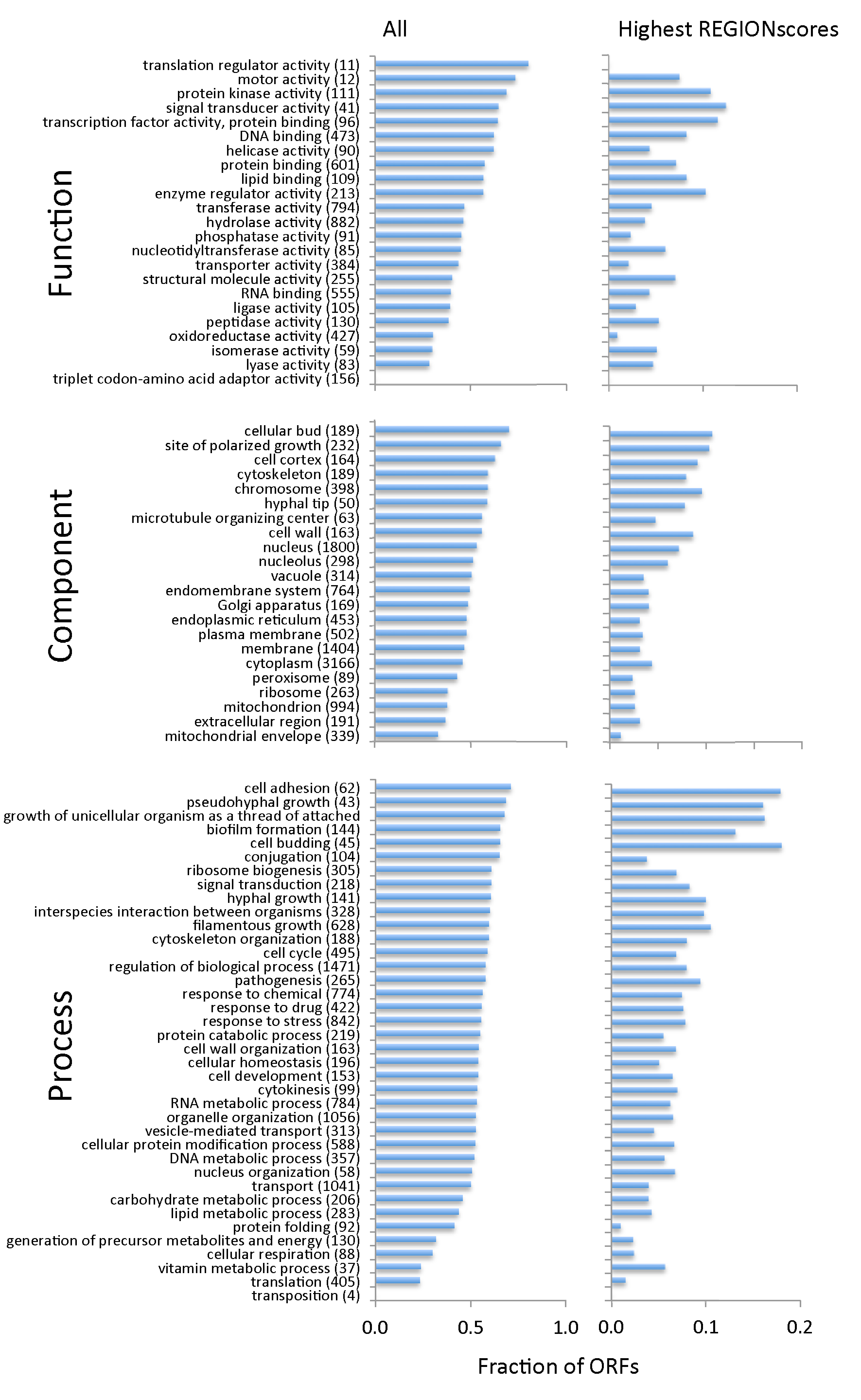
3.3. TR-ORFs Are Not Restricted to Particular Functions, Processes and Components
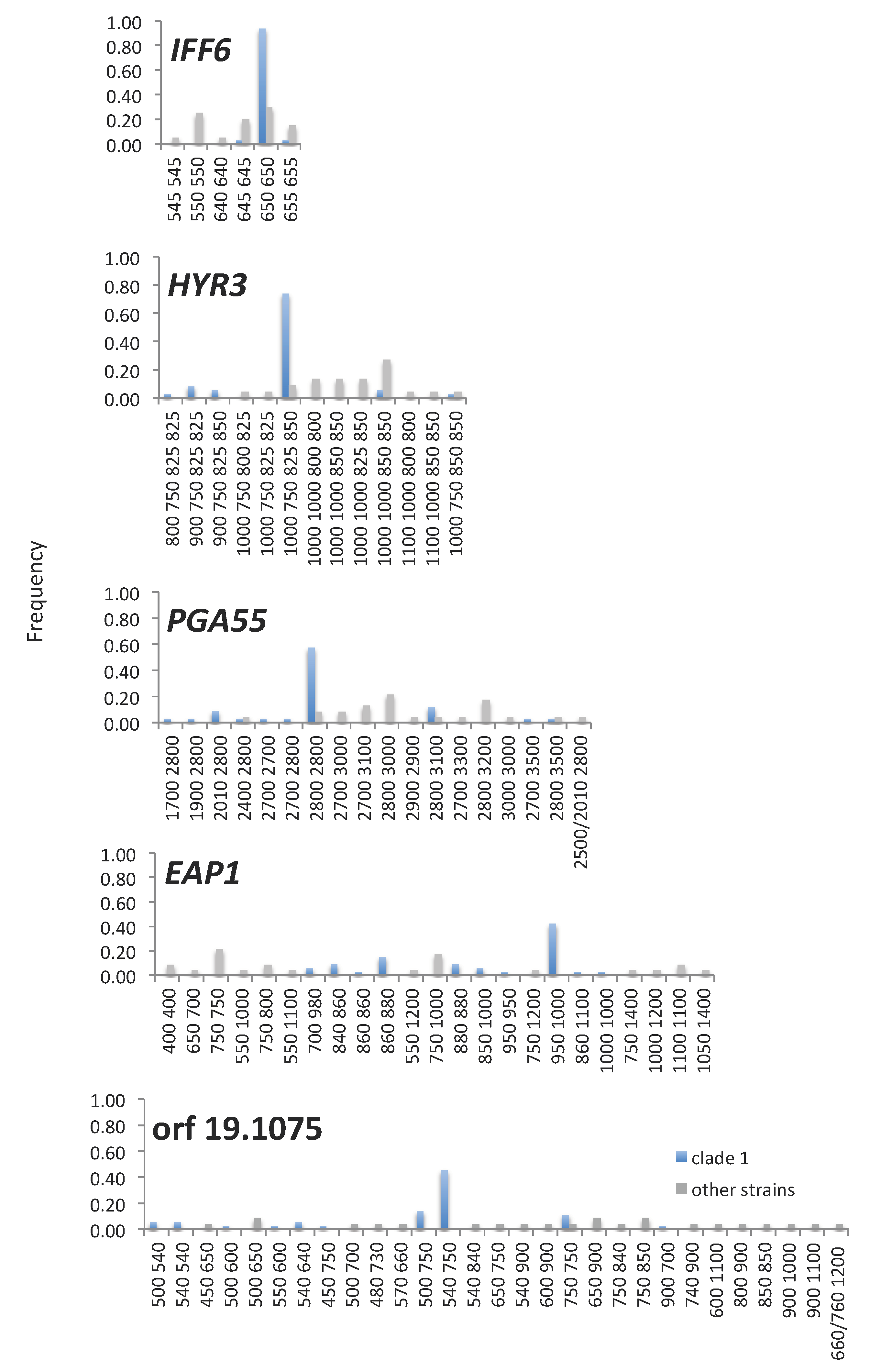
3.4. Amino Acid Repeat Units and Amino Acid Repeat Region Lengths Are under Selection and TR Mutation Generates Clade-Specific Protein Variants
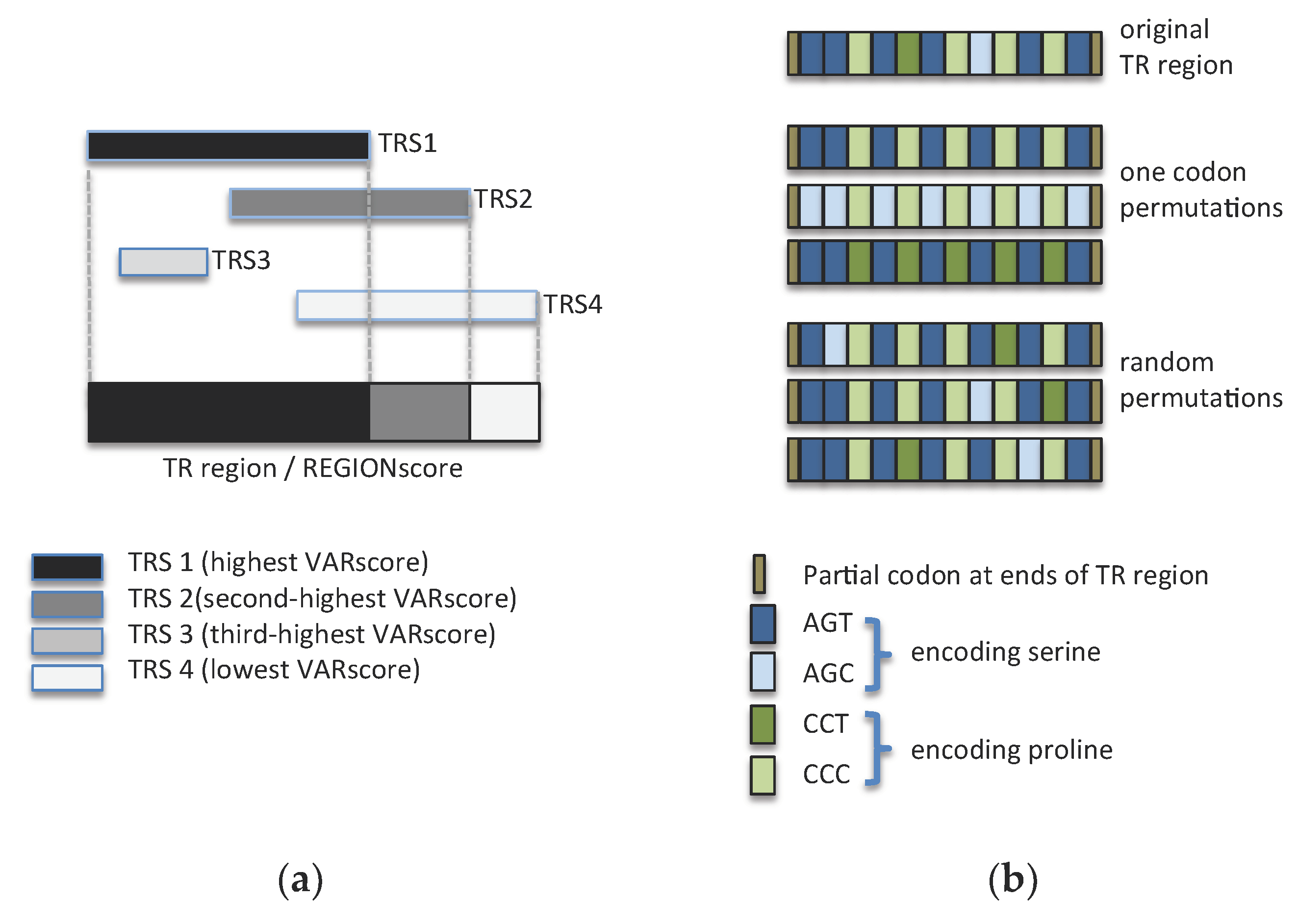
3.5. Synonymous Mutations Reduce the Repetitiveness of Many TR Regions
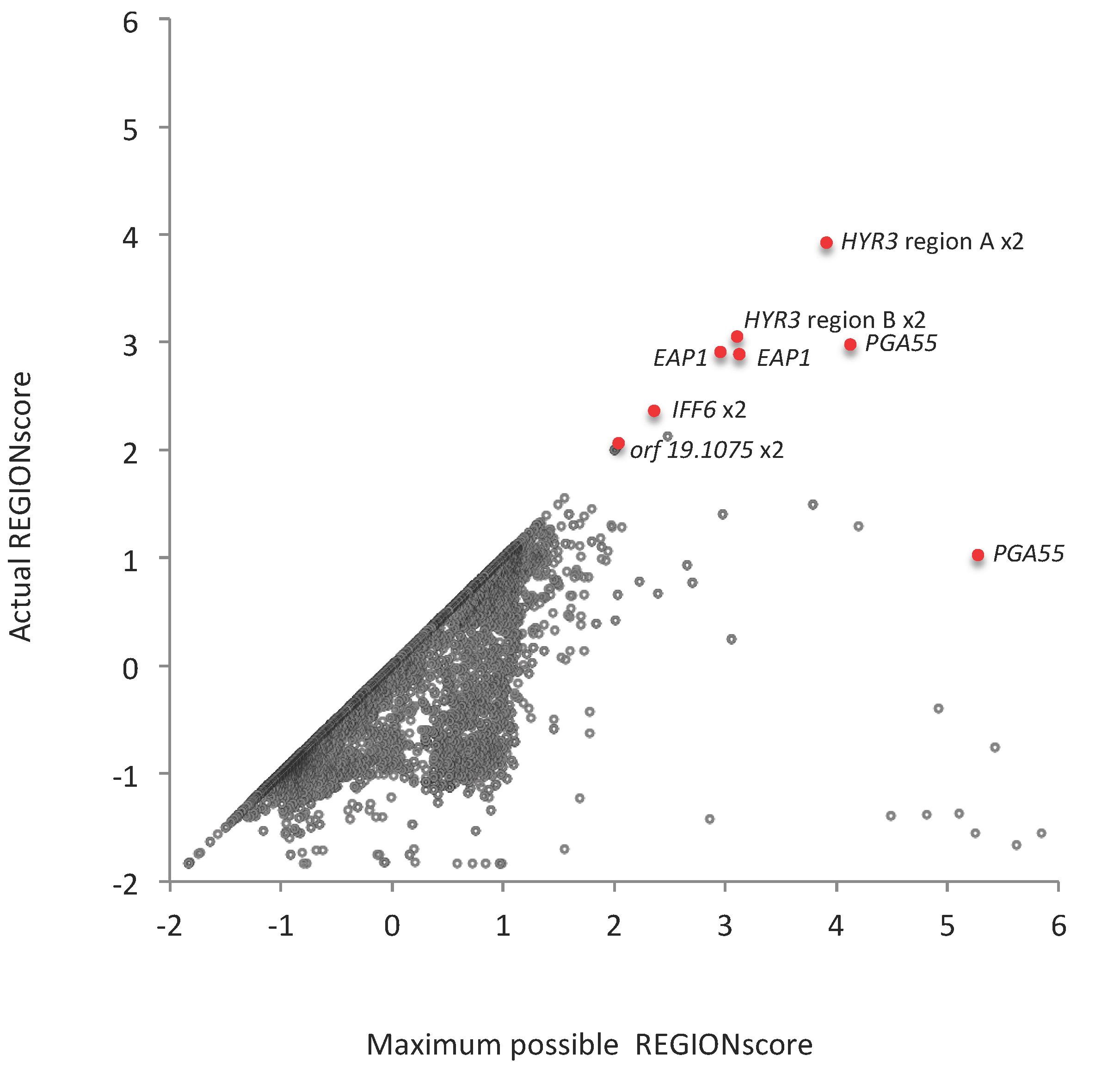
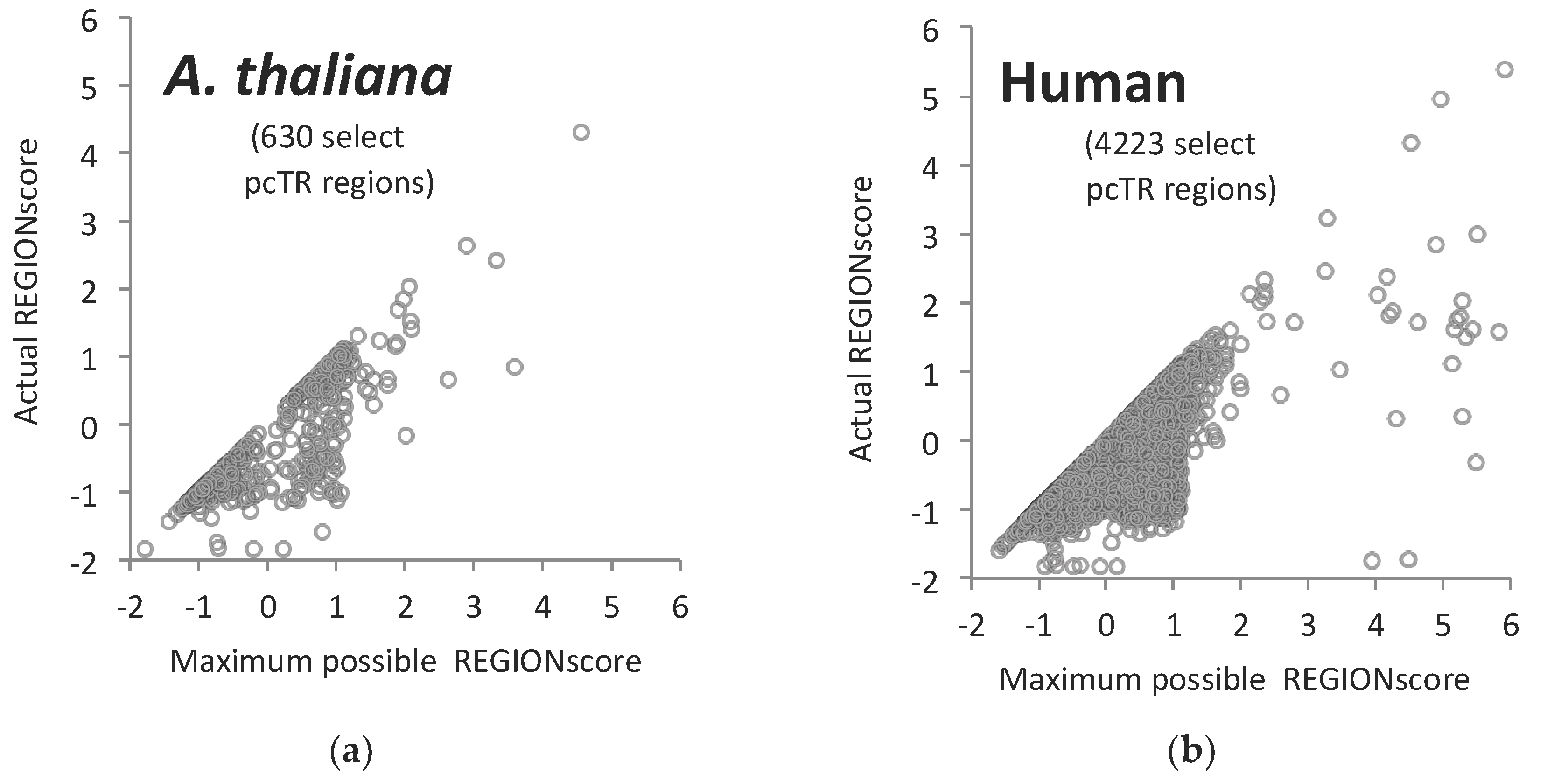
3.6. TR-ORFS with Close-to-Maximal REGION Scores. Are They Contingency Genes?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Liang, P. Transposable elements are a significant contributor to tandem repeats in the human genome. Comp. Funct. Genom. 2012, 2012, 947089. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.S.; Breden, F. Slipped-strand mispairing at noncontiguous repeats in poecilia reticulata: A model for minisatellite birth. Genetics 2000, 155, 1313–1320. [Google Scholar] [PubMed]
- Haber, J.E.; Louis, E.J. Minisatellite origins in yeast and humans. Genomics 1998, 48, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Cho, J.; Boeynaems, S.; Verstrepen, K.J. Beyond junk-variable tandem repeats as facilitators of rapid evolution of regulatory and coding sequences. Genes 2012, 3, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Chavali, S.; Pougach, K.; Legendre, M.; Zhu, B.; Boeynaems, S.; van der Zande, E.; Gevaert, K.; Rousseau, F.; Schymkowitz, J.; et al. Variable glutamine-rich repeats modulate transcription factor activity. Mol. Cell 2015, 59, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.; Gemayel, R.; Verstrepen, K.J. Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn. 2012, 7, 108–125. [Google Scholar] [PubMed]
- Deitsch, K.W.; Lukehart, S.A.; Stringer, J.R. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 2009, 7, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Hancock, J.M. Tandem and cryptic amino acid repeats accumulate in disordered regions of proteins. Genome Biol. 2009, 10, R59. [Google Scholar] [CrossRef] [PubMed]
- Albà, M.M.; Guigó, R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 2004, 14, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The origins of genome complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.J.; Bensasson, D.; Burt, A.; Koufopanou, V. Population genomics of the wild yeast Saccharomyces paradoxus: Quantifying the life cycle. Proc. Natl. Acad. Sci. USA 2008, 105, 4957–4962. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B. Fundamental concepts in genetics: Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Sim, K.L.; Creamer, T.P. Protein simple sequence conservation. Proteins 2004, 54, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.; Fundyga, R.; Hecht, J.E.; Kapteyn, J.C.; Klis, F.M.; Arnold, J. Characterization of agglutinin-like sequence genes from non-albicans Candida and phylogenetic analysis of the als family. Genetics 2001, 157, 1555–1567. [Google Scholar] [PubMed]
- Zhou, Z.; Jordens, Z.; Zhang, S.; Zhang, N.; Schmid, J. Highly mutable tandem DNA repeats generate a cell wall protein variant more frequent in disease-causing Candida albicans isolates than in commensal isolates. PLoS ONE 2017, 12, e0180246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Cannon, R.D.; Holland, B.; Patchett, M.; Schmid, J. Impact of genetic background on allele selection in a highly mutable Candida albicans gene, PNG2. PLoS ONE 2010, 5, e9614. [Google Scholar] [CrossRef]
- Zhou, K.; Aertsen, A.; Michiels, C.W. The role of variable DNA tandem repeats in bacterial adaptation. FEMS Microbiol. Rev. 2014, 38, 119–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.C. Candida and Candidosis, 2nd ed.; Bailliere Tindall: London, UK, 1988. [Google Scholar]
- Skinner, C.E.; Fletcher, D.W. A review of the genus Candida. Bacteriol. Rev. 1960, 24, 397–416. [Google Scholar] [PubMed]
- Braun, B.R.; van het Hoog, M.; d’Enfert, C.; Martchenko, M.; Dungan, J.; Kuo, A.; Inglis, D.O.; Uhl, M.A.; Hogues, H.; Berriman, M.; et al. A human-curated annotation of the Candida albicans genome. PLoS Genet. 2005, 1, 36–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, J.; Magee, P.T.; Holland, B.R.; Zhang, N.; Cannon, R.D.; Magee, B.B. Last hope for the doomed? Thoughts on the importance of a parasexual cycle for the yeast Candida albicans. Curr. Genet. 2015, 62, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.M.; Wang, Z.; Marjani, S.L.; Euskirchen, G.M.; Martin, J.; Sherlock, G.; Snyder, M. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010, 20, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.; Inglis, D.; Skrzypek, M.; Binkley, J.; Shah, P.; Wymore, F.; Binkley, G.; Miyasato, S.; Simison, M.; Sherlock, G. Candida Genome Database. Available online: http://www.candidagenome.org/ (accessed on 15 April 2018).
- Legendre, M.; Pochet, N.; Pak, T.; Verstrepen, K.J. Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res. 2007, 17, 1787–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyse DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Holland, B.R.; Schmid, J. Selecting representative model micro-organisms. BMC Microbiol. 2005, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, J.; Voss, E.; Soll, D.R. Computer-assisted methods for assessing strain relatedness in Candida albicans by fingerprinting with the moderately repetitive sequence Ca3. J. Clin. Microbiol. 1990, 28, 1236–1243. [Google Scholar] [PubMed]
- Soll, D.R.; Galask, R.; Schmid, J.; Hanna, C.; Mac, K.; Morrow, B. Genetic dissimilarity of commensal strains of Candida spp. Carried in different anatomical locations of the same healthy women. J. Clin. Microbiol. 1991, 29, 1702–1710. [Google Scholar] [PubMed]
- Schmid, J.; Odds, F.C.; Wiselka, M.J.; Nicholson, K.G.; Soll, D.R. Genetic similarity and maintenance of Candida albicans strains from a group of aids patients, demonstrated by DNA fingerprinting. J. Clin. Microbiol. 1992, 30, 935–941. [Google Scholar] [PubMed]
- Schmid, J.; Rotman, M.; Reed, B.; Pierson, C.L.; Soll, D.R. Genetic similarity of Candida albicans strains from vaginitis patients and their partners. J. Clin. Microbiol. 1993, 31, 39–46. [Google Scholar] [PubMed]
- Schmid, J.; Tay, Y.P.; Wan, L.; Carr, M.; Parr, D.; McKinney, W. Evidence for nosocomial transmission of Candida albicans obtained by ca3 fingerprinting. J. Clin. Microbiol. 1995, 33, 1223–1230. [Google Scholar] [PubMed]
- Schmid, J.; Herd, S.; Hunter, P.R.; Cannon, R.D.; Yasin, M.S.M.; Samad, S.; Carr, M.; Parr, D.; McKinney, W.; Schousboe, M.; et al. Evidence for a general-purpose genotype in Candida albicans, highly prevalent in multiple geographic regions, patient types and types of infection. Microbiology 1999, 145, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Sung, W.; Morris, K.; Coffey, N.; Landry, C.R.; Dopman, E.B.; Dickinson, W.J.; Okamoto, K.; Kulkarni, S.; Hartl, D.L.; et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl. Acad. Sci. USA 2008, 105, 9272–9277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Raja, J.; Larriba, G. Comparison of two approaches for identification of haplotypes and point mutations in Candida albicans and saccharomyces cerevisiae. J. Microbiol. Methods 2013, 94, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, T.; Faiza, M.; Pant, P.; Rayyan Akhtar, M.; Pant, P. An analysis of single nucleotide substitution in genetic codons—probabilities and outcomes. Bioinformation 2016, 12, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Young, E.T.; Sloan, J.S.; Van Riper, K. Trinucleotide repeats are clustered in regulatory genes in saccharomyces cerevisiae. Genetics 2000, 154, 1053–1068. [Google Scholar] [PubMed]
- Staab, J.F.; Bahn, Y.S.; Tai, C.H.; Cook, P.F.; Sundstrom, P. Expression of transglutaminase substrate activity on Candida albicans germ tubes through a coiled, disulfide-bonded n-terminal domain of Hwp1 requires c-terminal glycosylphosphatidylinositol modification. J. Biol. Chem. 2004, 279, 40737–40747. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Palecek, S.P. Distinct domains of the Candida albicans adhesin Eap1p mediate cell-cell and cell-substrate interactions. Microbiology 2008, 154, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.; Lai, W.-C.; Lee, T.-L.; Tseng, T.-L.; Shieh, J.-C. Dissection of the Candida albicans Cdc4 protein reveals the involvement of domains in morphogenesis and cell flocculation. J. Biomed. Sci. 2013, 20, 97. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, X.; Ding, Y.; Chen, J. Asc1, a wd-repeat protein, is required for hyphal development and virulence in Candida albicans. Acta Biochim. Biophys. Sin. 2010, 42, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Boisrame, A.; Cornu, A.; Da Costa, G.; Richard, M.L. Unexpected role for a serine/threonine-rich domain in the Candida albicans Iff protein family. Eukaryot. Cell 2011, 10, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-H.; Cheng, G.; Nuessen, J.A.; Jajko, R.; Yeater, K.M.; Zhao, X.; Pujol, C.; Soll, D.R.; Hoyer, L.L. Functional specificity of Candida albicans Als3p proteins and clade specificity of ALS3 alleles discriminated by the number of copies of the tandem repeat sequence in the central domain. Microbiology 2005, 151, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Rauceo, J.M.; De Armond, R.; Otoo, H.; Kahn, P.C.; Klotz, S.A.; Gaur, N.K.; Lipke, P.N. Threonine-rich repeats increase fibronectin binding in the Candida albicans adhesin Als5p. Eukaryot. Cell 2006, 5, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Sumita, T.; Yoko-o, T.; Shimma, Y.-i.; Jigami, Y. Comparison of cell wall localization among pir family proteins and functional dissection of the region required for cell wall binding and bud scar recruitment of Pir1p. Eukaryot. Cell 2005, 4, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, P.; Nogueira, E.; Loureiro, A.; Delgado-Silva, Y.; Correia, A.; Pais, C. Increased number of glutamine repeats in the c-terminal of Candida albicans Rlm1p enhances the resistance to stress agents. Antonie Van Leeuwenhoek 2009, 96, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Harrex, A.L.; Holland, B.R.; Fenton, L.E.; Cannon, R.D.; Schmid, J. Sixty alleles of the ALS7 open reading frame in Candida albicans: Als7 is a hypermutable contingency locus. Genome Res. 2003, 13, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Lott, T.J.; Fundyga, R.E.; Kuykendall, R.J.; Arnold, J. The human commensal yeast, Candida albicans, has an ancient origin. Fungal Genet. Biol. 2005, 42, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Lott, T.J.; Holloway, B.P.; Logan, D.A.; Fundyga, R.; Arnold, J. Towards understanding the evolution of the human commensal yeast Candida albicans. Microbiology 1999, 145, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Oh, S.H.; Jajko, R.; Diekema, D.J.; Pfaller, M.A.; Pujol, C.; Soll, D.R.; Hoyer, L.L. Analysis of ALS5 and ALS6 allelic variability in a geographically diverse collection of Candida albicans isolates. Fungal Genet. Biol. 2007, 44, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, D.M.; Castillo, L.; Nather, K.; Munro, C.A.; Brown, A.J.; Gow, N.A.; Odds, F.C. Property differences among the four major Candida albicans strain clades. Eukaryot. Cell. 2009, 8, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Upritchard, J.E.; Holland, B.R.; Fenton, L.E.; Ferguson, M.M.; Cannon, R.D.; Schmid, J. Distribution of mutations distinguishing the most prevalent disease-causing Candida albicans genotype from other genotypes. Infect. Genet. Evol. 2009, 9, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Ridley, M. Evolution; Blackwell Science: Cambridge, MA, USA, 1996. [Google Scholar]
- Tavanti, A.; Davidson, A.D.; Fordyce, M.J.; Gow, N.A.R.; Maiden, M.C.J.; Odds, F.C. Population structure and properties of Candida albicans, as determined by multilocus sequence typing. J. Clin. Microbiol. 2005, 43, 5601–5613. [Google Scholar] [CrossRef] [PubMed]
- Eyre-Walker, A.; Keightley, P.D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 2007, 8, 610. [Google Scholar] [CrossRef] [PubMed]
- Wloch, D.M.; Szafraniec, K.; Borts, R.H.; Korona, R. Direct estimate of the mutation rate and the distribution of fitness effects in the yeast Saccharomyces cerevisiae. Genetics 2001, 159, 441–452. [Google Scholar] [PubMed]
- Bougnoux, M.E.; Tavanti, A.; Bouchier, C.; Gow, N.A.; Magnier, A.; Davidson, A.D.; Maiden, M.C.; D’Enfert, C.; Odds, F.C. Collaborative consensus for optimized multilocus sequence typing of Candida albicans. J. Clin. Microbiol. 2003, 41, 5265–5266. [Google Scholar] [CrossRef] [PubMed]
- Forche, A.; Magee, P.T.; Magee, B.B.; May, G. Genome-wide single-nucleotide polymorphism map for Candida albicans. Eukaryot. Cell 2004, 3, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol Cell. Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.; Tortorano, A.M.; Jones, G.; Lazzarini, C.; Zhang, N.; Bendall, M.J.; Cogliati, M.; Wattimena, S.; Klingspor, L.; ECMM_survey_participants; et al. Increased mortality in young candidemia patients associated with presence of a Candida albicans general-purpose genotype. J. Clin. Microbiol. 2011, 49, 3250–3256. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C.; Bougnoux, M.E.; Shaw, D.J.; Bain, J.M.; Davidson, A.D.; Diogo, D.; Jacobsen, M.D.; Lecomte, M.; Li, S.Y.; Tavanti, A.; et al. Molecular phylogenetics of Candida albicans. Eukaryot. Cell 2007, 6, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wheeler, D.; Truglio, M.; Lazzarini, C.; Upritchard, J.; McKinney, W.; Rogers, K.; Prigitano, A.; Tortorano, A.M.; Cannon, R.D.; et al. Multi-locus next-generation sequence typing of DNA extracted from pooled colonies detects multiple unrelated Candida albicans strains in a significant proportion of patient samples. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Magee, B.B.; Magee, P.T.; Holland, B.R.; Rodrigues, E.; Holmes, A.R.; Cannon, R.D.; Schmid, J. Selective advantages of a parasexual cycle for the yeast Candida albicans. Genetics 2015, 200, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Massicotte, R.; Angers, B. General-purpose genotype or how epigenetics extend the flexibility of a genotype. Genet. Res. Int. 2012, 2012, 317175. [Google Scholar] [CrossRef] [PubMed]
- Stearns, S.C. The evolutionary significance of phenotypic plasticityphenotypic sources of variation among organisms can be described by developmental switches and reaction norms. Bioscience 1989, 39, 436–445. [Google Scholar] [CrossRef]
- Moxon, R.; Bayliss, C.; Hood, D. Bacterial contingency loci: The role of simple sequence DNA repeats in bacterial adaptation. Annu. Rev. Genet. 2006, 40, 307–333. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, K.J.; Reynolds, T.B.; Fink, G.R. Origins of variation in the fungal cell surface. Nat. Rev. Microbiol. 2004, 2, 533–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virji, M. Pathogenic neisseriae: Surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 2009, 7, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Oh, S.H.; Hoyer, L.L. Unequal contribution of ALS9 alleles to adhesion between Candida albicans and human vascular endothelial cells. Microbiology 2007, 153, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.L.; Scherer, S.; Shatzman, A.R.; Livi, G.P. Candida albicans ALS1: Domains related to a Saccharomyces cerevisiae sexual agglutinin separated by a repeating motif. Mol. Microbiol. 1995, 15, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L. The als gene family of Candida albicans. Trends Microbiol. 2001, 9, 176–180. [Google Scholar] [CrossRef]
- Richardson, J.P.; Moyes, D.L. Adaptive immune responses to Candida albicans infection. Virulence 2015, 6, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, K.J.; Fink, G.R. Genetic and epigenetic mechanisms underlying cell-surface variability in protozoa and fungi. Annu. Rev. Genet. 2009, 43, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Perez, J.I.; Gonzalez, A.A.; Toranzos, G.A. From evolutionary advantage to disease agents: Forensic reevaluation of host-microbe interactions and pathogenicity. Microbiol. Spectrum 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Diekema, D.J.; Messer, S.A.; Brueggemann, A.B.; Coffman, S.L.; Doern, G.V.; Herwaldt, L.A.; Pfaller, M.A. Epidemiology of candidemia: 3-year results from the emerging infections and the epidemiology of Iowa organisms study. J. Clin. Microbiol. 2002, 40, 1298–1302. [Google Scholar] [CrossRef] [PubMed]
- Filippidi, A.; Galanakis, E.; Maraki, S.; Galani, I.; Drogari-Apiranthitou, M.; Kalmanti, M.; Mantadakis, E.; Samonis, G. The effect of maternal flora on Candida colonisation in the neonate. Mycoses 2014, 57, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Brockhurst, M.A.; Chapman, T.; King, K.C.; Mank, J.E.; Paterson, S.; Hurst, G.D. Running with the red queen: The role of biotic conflicts in evolution. Proc. Biol. Sci. 2014, 281, 20141382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomas, W.K.; Foster, P.L. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Au, A.Y.; Ritchie, W.; Rasko, J.E. Intron retention in mRNA: No longer nonsense: Known and putative roles of intron retention in normal and disease biology. Bioessays 2016, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sznajder, Ł.J.; Thomas, J.D.; Carrell, E.M.; Reid, T.; McFarland, K.N.; Cleary, J.D.; Oliveira, R.; Nutter, C.A.; Bhatt, K.; Sobczak, K.; et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. USA 2018, 115, 4234. [Google Scholar] [CrossRef] [PubMed]
- Matoulkova, E.; Michalova, E.; Vojtesek, B.; Hrstka, R. The role of the 3’ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012, 9, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TR Region 1 | REGION Score | TR Unit Lengths (bp) 2 | Nucleic Acid and Amino Acid Sequence of Regions 3 |
|---|---|---|---|
| C2_10280C_B 3 (SPT23) | 1.01 | 3 | ACAACAACAACAACAACAACAACAACAACAACAACAACAGCAGCAGCAACAGCAACAACAGCAA |
| QQQQQQQQQQQQQQQQQQQQQ | |||
| C1_01560W_B 1 (SIZ1) | 0.92 | 6, 3, 12, 24 | ACAGCAACAGCAACAGCGACAACTTCGACAACTAGAACAGCAGCAACGGCTACAGCGACAGCAATGGCAACAGCAACAGCAACAACTACAGCAACAACAACAACAACTTCCTCGCCAACTTCCCCAACAGTCTCCCCGACAACTTCCCCAACAG |
| QQQQQRQLRQLEQQQRLQRQQWQQQQQQLQQQQQQLPRQLPQQSPRQLPQQ | |||
| C2_02330W_B 1 | 0.28 | 4 | CACTTACTCACTCACTCACTCACTCACTC |
| LTHSLTHSL | |||
| C1_09690W_B 2 | −1.15 | 12 | TTTCTTGCCAGATTTCTTACCAGA |
| (MLS1) | FLPDFLP |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkins, M.; Zhang, N.; Schmid, J. Biological Roles of Protein-Coding Tandem Repeats in the Yeast Candida Albicans. J. Fungi 2018, 4, 78. https://doi.org/10.3390/jof4030078
Wilkins M, Zhang N, Schmid J. Biological Roles of Protein-Coding Tandem Repeats in the Yeast Candida Albicans. Journal of Fungi. 2018; 4(3):78. https://doi.org/10.3390/jof4030078
Chicago/Turabian StyleWilkins, Matt, Ningxin Zhang, and Jan Schmid. 2018. "Biological Roles of Protein-Coding Tandem Repeats in the Yeast Candida Albicans" Journal of Fungi 4, no. 3: 78. https://doi.org/10.3390/jof4030078
APA StyleWilkins, M., Zhang, N., & Schmid, J. (2018). Biological Roles of Protein-Coding Tandem Repeats in the Yeast Candida Albicans. Journal of Fungi, 4(3), 78. https://doi.org/10.3390/jof4030078