
Engineering and Implementation of Synthetic Molecular Tools in the Basidiomycete Fungus Ustilago maydis

 , and
, and {kind=link}
{kind=link}
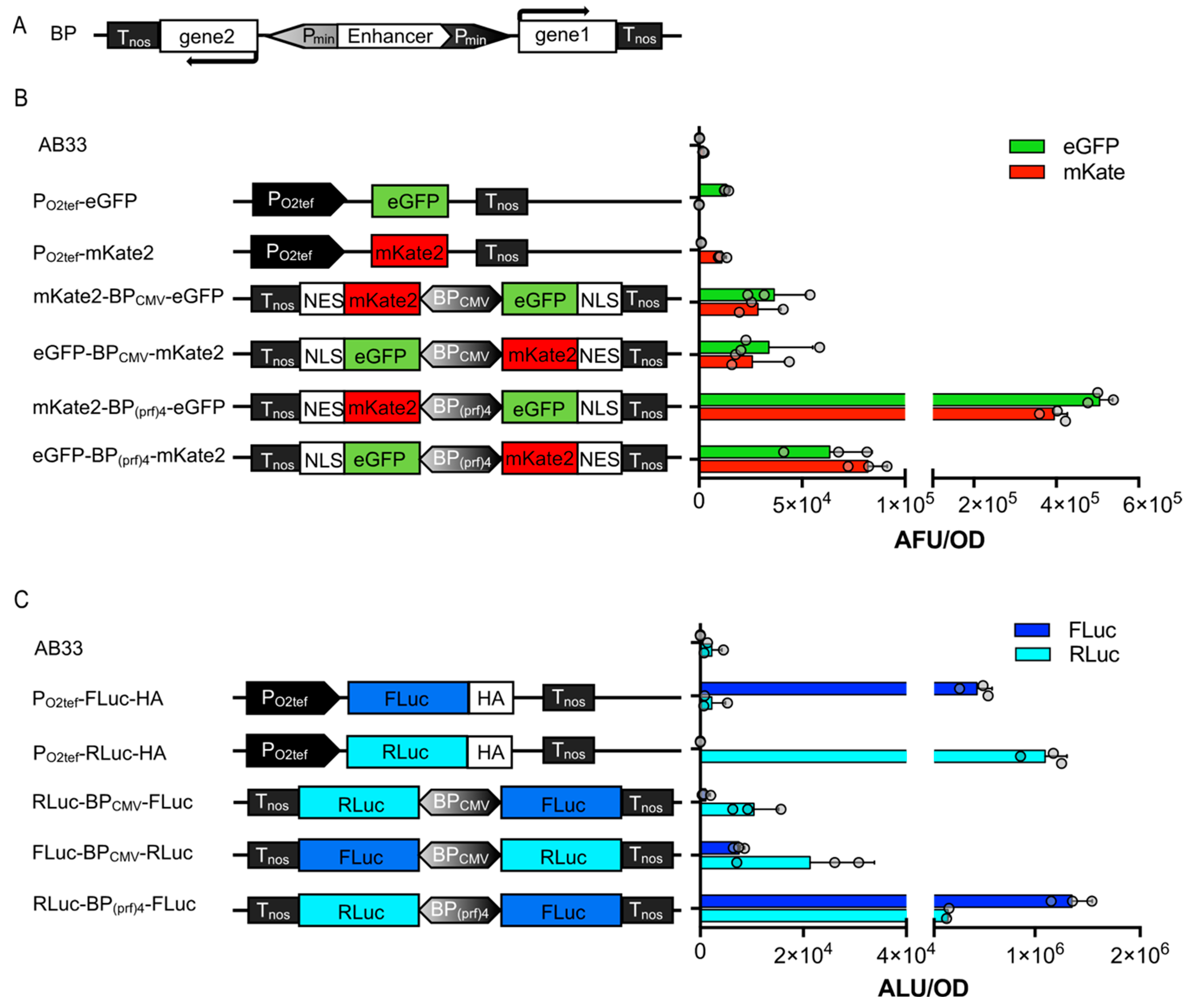{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Generation
2.2. Strain Generation and Growth Conditions
2.3. Luminescence Determination
2.4. SEAP Reporter Assay
2.5. Fluorescence Intensity Measurements
2.6. Induction of Filaments and Microscopy
2.7. Infection Assays and In Planta Luciferase Quantification
3. Results
3.1. Quantitative Reporter Gene Expression
3.2. Ratiometric Monitoring of Inducible Gene Expression
3.3. Bidirectional Promoters for Bicistronic Expression in U. maydis
3.4. IRES Sequences
3.5. Quantitative Determination of Fungal Proliferation In Planta
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldbrügge, M.; Kellner, R.; Schipper, K. The biotechnological use and potential of plant pathogenic smut fungi. Appl. Microbiol. Biotechnol. 2013, 97, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Banuett, F. Ustilago maydis, the delightful blight. Trends Genet. 1992, 8, 174–180. [Google Scholar] [CrossRef]
- Olicón-Hernández, D.R.; Araiza-Villanueva, M.G.; Pardo, J.P.; Aranda, E.; Guerra-Sánchez, G. New Insights of Ustilago maydis as Yeast Model for Genetic and Biotechnological Research: A Review. Curr. Microbiol. 2019, 76, 917–926. [Google Scholar] [CrossRef]
- Vollmeister, E.; Schipper, K.; Baumann, S.; Haag, C.; Pohlmann, T.; Stock, J.; Feldbrügge, M. Fungal development of the plant pathogen Ustilago maydis. FEMS Microbiol. Rev. 2012, 36, 59–77. [Google Scholar] [CrossRef]
- Zuo, W.; Ökmen, B.; Depotter, J.R.L.; Ebert, M.K.; Redkar, A.; Villamil, J.M.; Doehlemann, G. Molecular Interactions between Smut Fungi and Their Host Plants. Annu. Rev. Phytopathol. 2019, 57, 411–430. [Google Scholar] [CrossRef]
- Ferris, A.C.; Walbot, V. Understanding Ustilago maydis infection of multiple maize organs. J. Fungi 2021, 7, 8. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Brachmann, A.; König, J.; Julius, C.; Feldbrügge, M. A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol. Genet. Genom. 2004, 272, 216–226. [Google Scholar] [CrossRef]
- Terfrüchte, M.; Joehnk, B.; Fajardo-Somera, R.; Braus, G.H.; Riquelme, M.; Schipper, K.; Feldbrügge, M. Establishing a versatile Golden Gate cloning system for genetic engineering in fungi. Fungal Genet. Biol. 2014, 62, 1–10. [Google Scholar] [CrossRef]
- Lee, J.; Hilgers, F.; Loeschke, A.; Jaeger, K.E.; Feldbrügge, M. Ustilago maydis Serves as a Novel Production Host for the Synthesis of Plant and Fungal Sesquiterpenoids. Front. Microbiol. 2020, 11, 1655. [Google Scholar] [CrossRef]
- Wierckx, N.; Miebach, K.; Ihling, N.; Hussnaetter, K.P.; Büchs, J.; Schipper, K. Perspectives for the application of Ustilaginaceae as biotech cell factories. Essays Biochem. 2021, 65, 365–379. [Google Scholar] [CrossRef]
- Carstensen, F.; Klement, T.; Büchs, J.; Melin, T.; Wessling, M. Continuous production and recovery of itaconic acid in a membrane bioreactor. Bioresour. Technol. 2013, 137, 179–187. [Google Scholar] [CrossRef]
- Schuster, M.; Schweizer, G.; Reissmann, S.; Kahmann, R. Genome editing in Ustilago maydis using the CRISPR–Cas system. Fungal Genet. Biol. 2016, 89, 3–9. [Google Scholar] [CrossRef]
- Brachmann, A.; Weinzierl, G.; Kämper, J.; Kahmann, R. Identification of genes in the bW/bE regulatory cascade in Ustilago maydis. Mol. Microbiol. 2001, 42, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Müntjes, K.; Philipp, M.; Hüsemann, L.; Heucken, N.; Weidtkamp-Peters, S.; Schipper, K.; Zurbriggen, M.D.; Feldbrügge, M. Establishing Polycistronic Expression in the Model Microorganism Ustilago maydis. Front. Microbiol. 2020, 11, 1384. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Crooks, C.; Lamb, C. High-throughput quantitative luminescence assay of the growth in planta of Pseudomonas syringae chromosomally tagged with Photorhabdus luminescens luxCDABE. Plant J. 2008, 53, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Wend, S.; Dal Bosco, C.; Kämpf, M.M.; Ren, F.; Palme, K.; Weber, W.; Dovzhenko, A.; Zurbriggen, M.D. A quantitative ratiometric sensor for time-resolved analysis of auxin dynamics. Sci. Rep. 2013, 3, 2052. [Google Scholar] [CrossRef] [PubMed]
- Samodelov, S.L.; Beyer, H.M.; Guo, X.; Augustin, M.; Jia, K.P.; Baz, L.; Ebenhöh, O.; Beyer, P.; Weber, W.; Al-Babili, S.; et al. Strigoquant: A genetically encoded biosensor for quantifying Strigolactone activity and specificity. Sci. Adv. 2016, 2, e1601266. [Google Scholar] [CrossRef]
- Müller, K.; Engesser, R.; Metzger, S.; Schulz, S.; Kämpf, M.M.; Busacker, M.; Steinberg, T.; Tomakidi, P.; Ehrbar, M.; Nagy, F.; et al. A red/far-red light-responsive bi-stable toggle switch to control gene expression in mammalian cells. Nucleic Acids Res. 2013, 41, e77. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Kickenweiz, T.; Pitzer, J.; Sturmberger, L.; Weninger, A.; Biggs, B.W.; Köhler, E.M.; Baumschlager, A.; Fischer, J.E.; Hyden, P.; et al. Engineered bidirectional promoters enable rapid multi-gene co-expression optimization. Nat. Commun. 2018, 9, 3589. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Beyer, H.M.; Gonschorek, P.; Samodelov, S.L.; Meier, M.; Weber, W.; Zurbriggen, M.D. AQUA cloning: A versatile and simple enzyme-free cloning approach. PLoS ONE 2015, 10, e0137652. [Google Scholar] [CrossRef] [PubMed]
- Doehlemann, G.; Reissmann, S.; Aßmann, D.; Fleckenstein, M.; Kahmann, R. Two linked genes encoding a secreted effector and a membrane protein are essential for Ustilago maydis-induced tumour formation. Mol. Microbiol. 2011, 81, 751–766. [Google Scholar] [CrossRef]
- Holliday, R. Ustilago maydis. In Bacteria, Bacteriophages, and Fungi; King, R.C., Ed.; Springer: Boston, MA, USA, 1974; Volume 1, pp. 575–595. ISBN 978-1-4899-1710-2. [Google Scholar]
- Bösch, K.; Frantzeskakis, L.; Vraneš, M.; Kämper, J.; Schipper, K.; Göhre, V. Genetic manipulation of the plant pathogen Ustilago maydis to study fungal biology and plant microbe interactions. J. Vis. Exp. 2016, 2016, e54522. [Google Scholar] [CrossRef]
- Golonka, D.; Fischbach, P.; Jena, S.G.; Kleeberg, J.R.W.; Essen, L.O.; Toettcher, J.E.; Zurbriggen, M.D.; Möglich, A. Deconstructing and repurposing the light-regulated interplay between Arabidopsis phytochromes and interacting factors. Commun. Biol. 2019, 2, 448. [Google Scholar] [CrossRef] [PubMed]
- Baumann, S.; Ko’nig, J.; Koepke, J.; Feldbrügge, M. Endosomal transport of septin mRNA and protein indicates local translation on endosomes and is required for correct septin filamentation. EMBO Rep. 2014, 15, 94–102. [Google Scholar] [CrossRef]
- Gelmini, S.; Pinzani, P.; Pazzagli, M. Luciferase gene as reporter: Comparison with the CAT gene and use in transfection and microinjection of mammalian cells. Methods Enzymol. 2000, 305, 557–576. [Google Scholar] [CrossRef]
- Srikantha, T.; Klapach, A.; Lorenz, W.W.; Tsai, L.K.; Laughlin, L.A.; Gorman, J.A.; Soll, D.R. The sea pansy Renilla reniformis luciferase serves as a sensitive bioluminescent reporter for differential gene expression in Candida albicans. J. Bacteriol. 1996, 178, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Badr, C.; Pike, L.; de Kleine, R.; Weissleder, R.; Breakefield, X.O.; Tannous, B.A. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 2008, 5, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.R.; Trotter, K.M.; Granados, R.R.; Wood, H.A. Baculovirus expression of alkaline phosphatase as a reporter gene for evaluation of production, glycosylation and secretion. Bio/Technology 1992, 10, 1148–1150. [Google Scholar] [CrossRef]
- Baaske, J.; Gonschorek, P.; Engesser, R.; Dominguez-Monedero, A.; Raute, K.; Fischbach, P.; Müller, K.; Cachat, E.; Schamel, W.W.A.; Minguet, S.; et al. Dual-controlled optogenetic system for the rapid down-regulation of protein levels in mammalian cells. Sci. Rep. 2018, 8, 15024. [Google Scholar] [CrossRef]
- Braguy, J.; Samodelov, S.L.; Andres, J.; Ochoa-Fernandez, R.; Al-Babili, S.; Zurbriggen, M.D. A Protoplast-Based Bioassay to Quantify Strigolactone Activity in Arabidopsis Using StrigoQuant. In Strigolactones: Methods and Protocols; Prandi, C., Cardinale, F., Eds.; Springer: New York, NY, USA, 2021; pp. 201–218. ISBN 978-1-0716-1429-7. [Google Scholar]
- Duttke, S.H.C.; Lacadie, S.A.; Ibrahim, M.M.; Glass, C.K.; Corcoran, D.L.; Benner, C.; Heinz, S.; Kadonaga, J.T.; Ohler, U. Human promoters are intrinsically directional. Mol. Cell 2015, 57, 674–684. [Google Scholar] [CrossRef]
- Sarkari, P.; Reindl, M.; Stock, J.; Müller, O.; Kahmann, R.; Feldbrügge, M.; Schipper, K. Improved expression of single-chain antibodies in Ustilago maydis. J. Biotechnol. 2014, 191, 165–175. [Google Scholar] [CrossRef]
- Hartmann, H.A.; Krüger, J.; Lottspeich, F.; Kahmann, R.; The, S.; Cell, P.; Jul, N.; Hartmann, H.A.; Kruger, J.; Lottspeich, F.; et al. Environmental Signals Controlling Sexual Development of the Corn Smut Fungus Ustilago maydis through the Transcriptional Regulator Prf1. Plant Cell 1999, 11, 1293–1305. [Google Scholar] [CrossRef]
- Andersen, C.R.; Nielsen, L.S.; Baer, A.; Tolstrup, A.B.; Weilguny, D. Efficient expression from one CMV enhancer controlling two core promoters. Mol. Biotechnol. 2011, 48, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Maurer, S.; Kaffarnik, F.; Ladendorf, O.; Brachmann, A.; Kämper, J.; Feldbrügge, M. Tetracycline-regulated gene expression in the pathogen Ustilago maydis. Fungal Genet. Biol. 2006, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Lanver, D.; Müller, A.N.; Happel, P.; Schweizer, G.; Haas, F.B.; Franitza, M.; Pellegrin, C.; Reissmann, S.; Altmüller, J.; Rensing, S.A.; et al. The biotrophic development of Ustilago maydis studied by RNA-seq analysis. Plant Cell 2018, 30, 300–323. [Google Scholar] [CrossRef] [PubMed]
- Frantzeskakis, L.; Courville, K.J.; Plücker, L.; Kellner, R.; Kruse, J.; Brachmann, A.; Feldbrügge, M.; Göhre, V. The plant-dependent life cycle of Thecaphora thlaspeos: A smut fungus adapted to brassicaceae. Mol. Plant-Microbe Interact. 2017, 30, 271–282. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heucken, N.; Tang, K.; Hüsemann, L.; Heßler, N.; Müntjes, K.; Feldbrügge, M.; Göhre, V.; Zurbriggen, M.D. Engineering and Implementation of Synthetic Molecular Tools in the Basidiomycete Fungus Ustilago maydis. J. Fungi 2023, 9, 480. https://doi.org/10.3390/jof9040480
Heucken N, Tang K, Hüsemann L, Heßler N, Müntjes K, Feldbrügge M, Göhre V, Zurbriggen MD. Engineering and Implementation of Synthetic Molecular Tools in the Basidiomycete Fungus Ustilago maydis. Journal of Fungi. 2023; 9(4):480. https://doi.org/10.3390/jof9040480
Chicago/Turabian StyleHeucken, Nicole, Kun Tang, Lisa Hüsemann, Natascha Heßler, Kira Müntjes, Michael Feldbrügge, Vera Göhre, and Matias D. Zurbriggen. 2023. "Engineering and Implementation of Synthetic Molecular Tools in the Basidiomycete Fungus Ustilago maydis" Journal of Fungi 9, no. 4: 480. https://doi.org/10.3390/jof9040480
APA StyleHeucken, N., Tang, K., Hüsemann, L., Heßler, N., Müntjes, K., Feldbrügge, M., Göhre, V., & Zurbriggen, M. D. (2023). Engineering and Implementation of Synthetic Molecular Tools in the Basidiomycete Fungus Ustilago maydis. Journal of Fungi, 9(4), 480. https://doi.org/10.3390/jof9040480