
Genomic and Transcriptomic Approaches Provide a Predictive Framework for Sesquiterpenes Biosynthesis in Desarmillaria tabescens CPCC 401429
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Cultivation Conditions
2.2. Sequencing, De Novo Assembly, and Bioinformatic Tools
2.3. RNA Purification, Library Construction, and RNA-Sequencing
2.4. Quality Control, Read Mapping, and Transcriptome Analysis
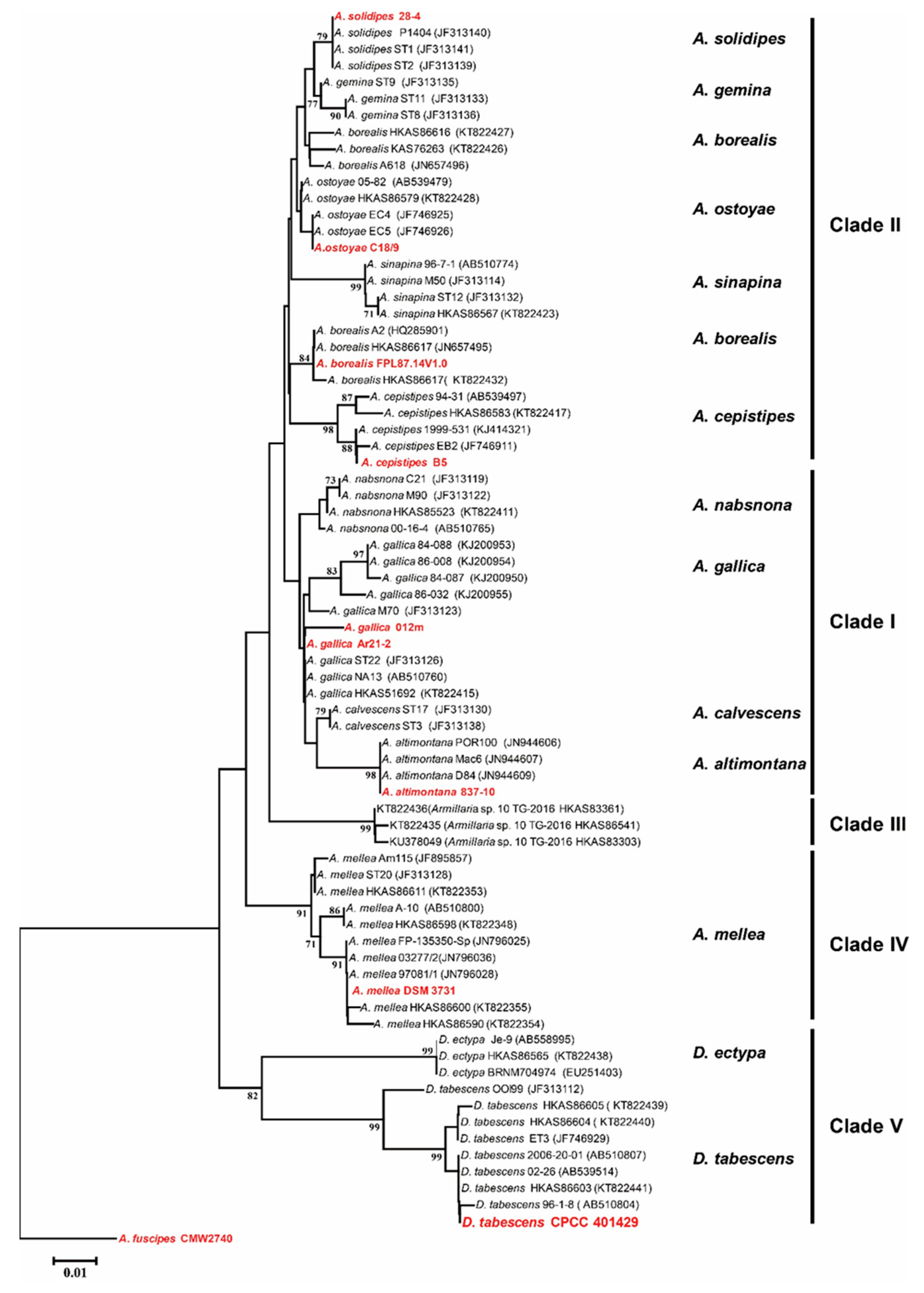
2.5. Multi-Locus Sequence Typing (MLST)-Based Phylogenetic Analysis
2.6. Bioinformatic Annotation for STSs and Phylogenetic Tree Construction
2.7. Gene Synthesis, Cloning and Expression of STSs in Yeast
2.8. GC-MS Analysis of Terpenoids
3. Results
3.1. General Genome Features of D. tabescens CPCC 401429
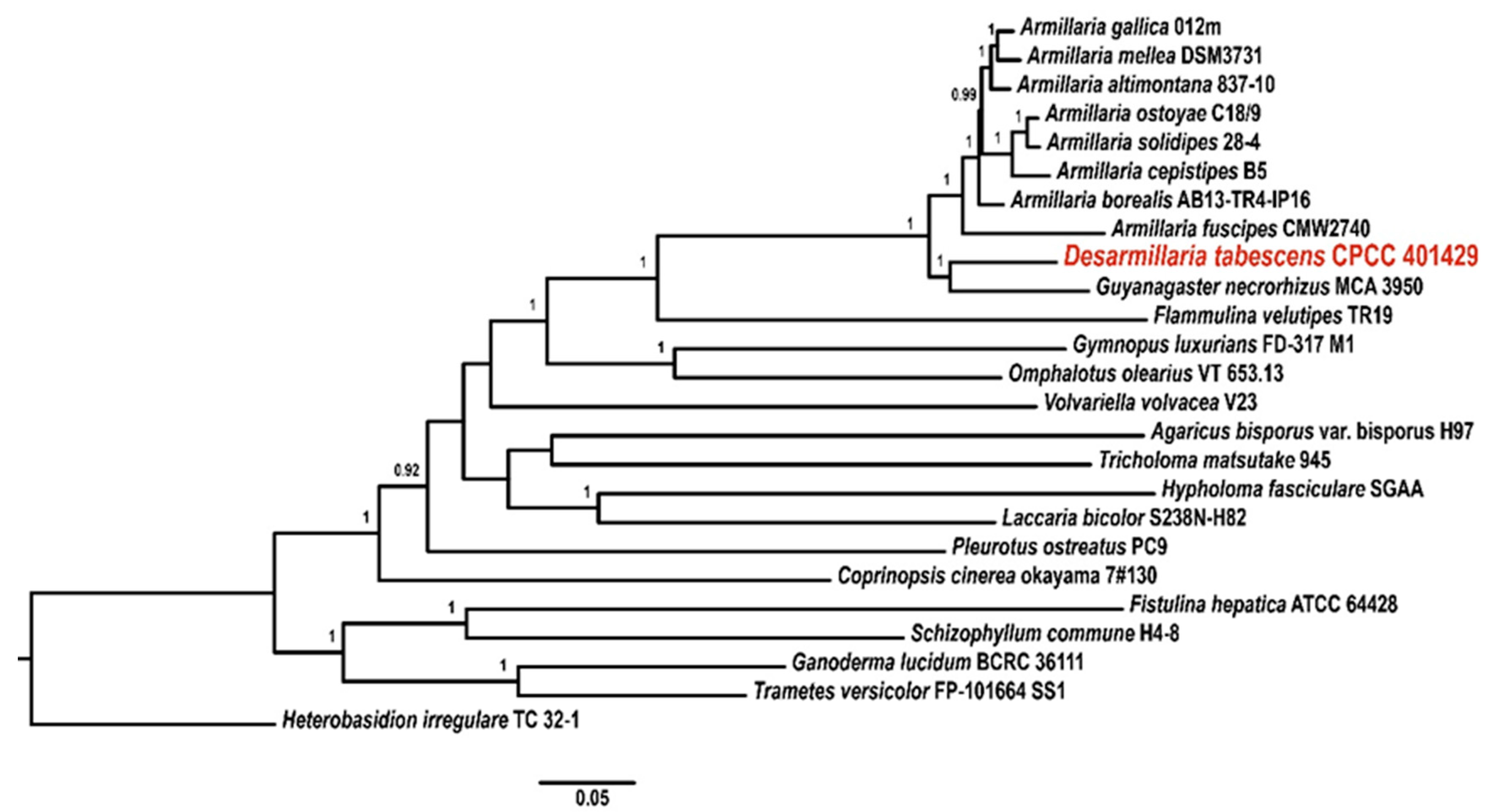
3.2. Phylogenetic Analysis of the Strain CPCC 401429
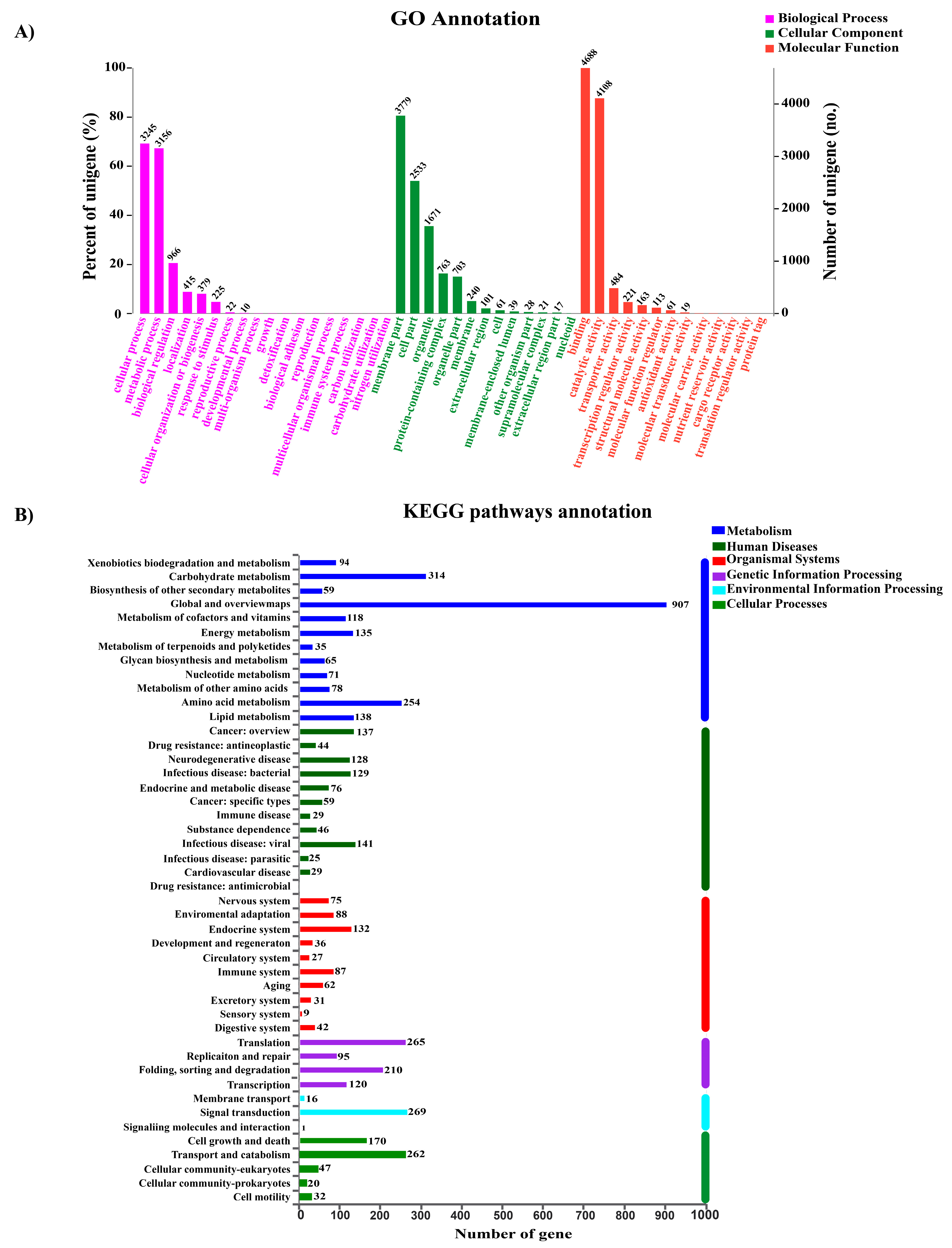
3.3. Genome Annotation and Bioinformatic Analysis
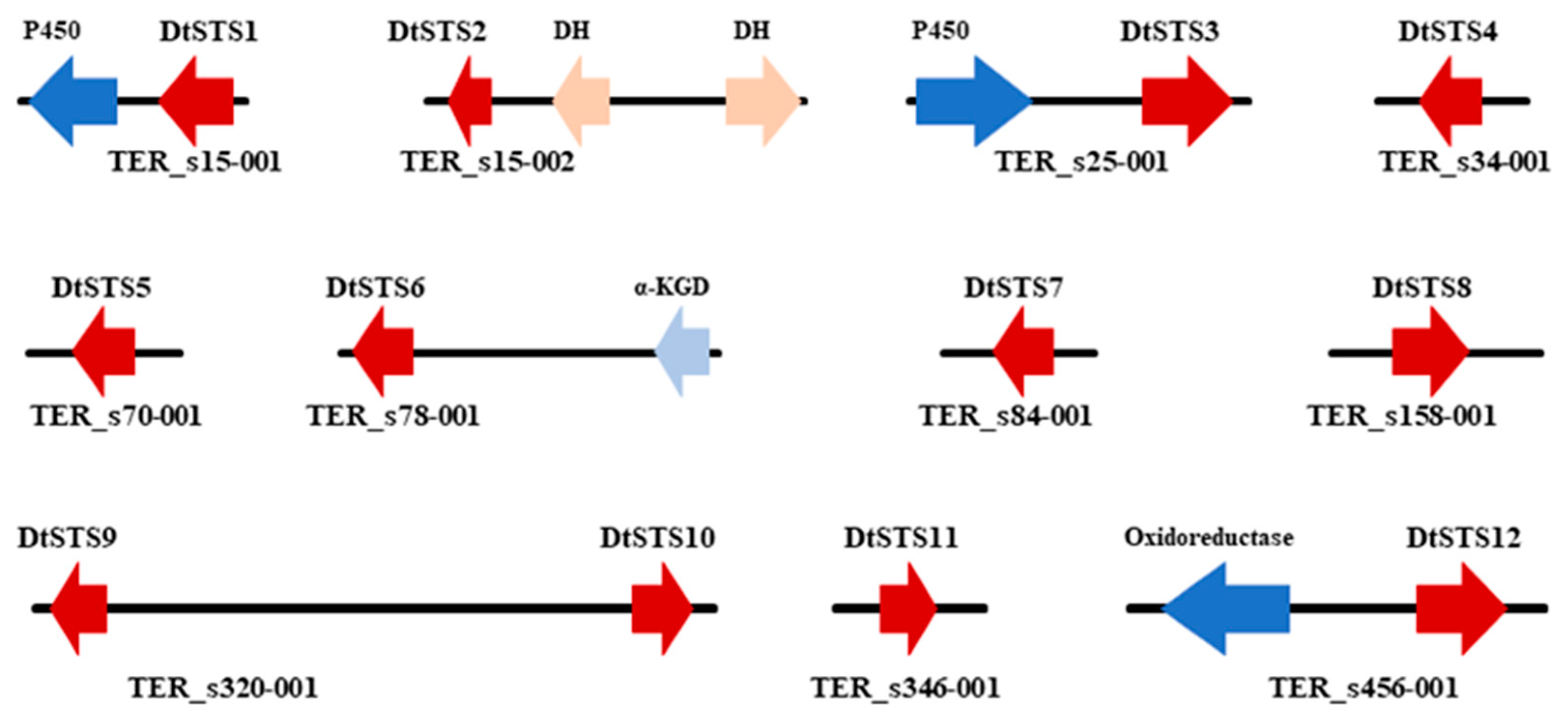
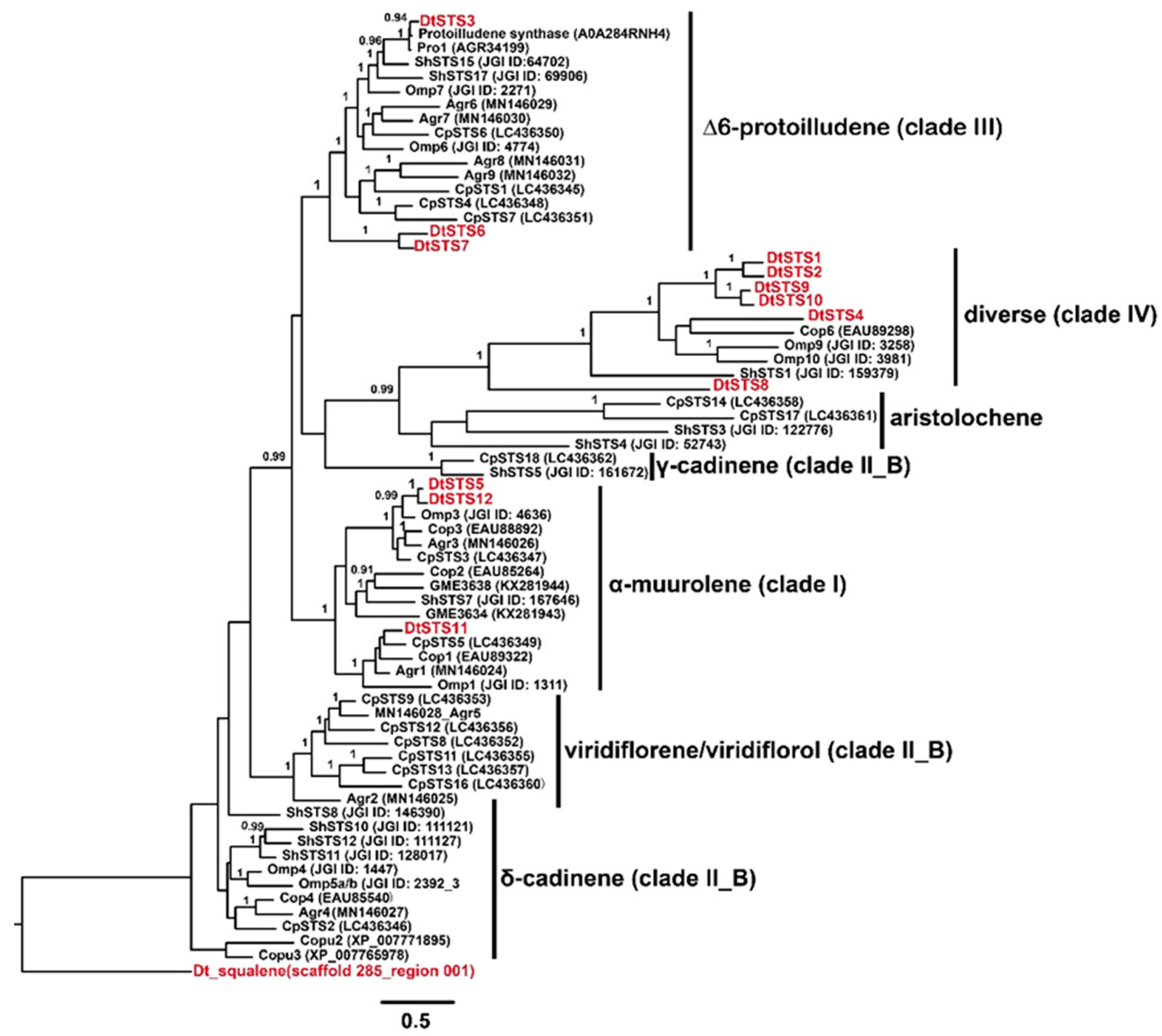
3.4. The Composition of STSs Homologues and Phylogeny of STSs
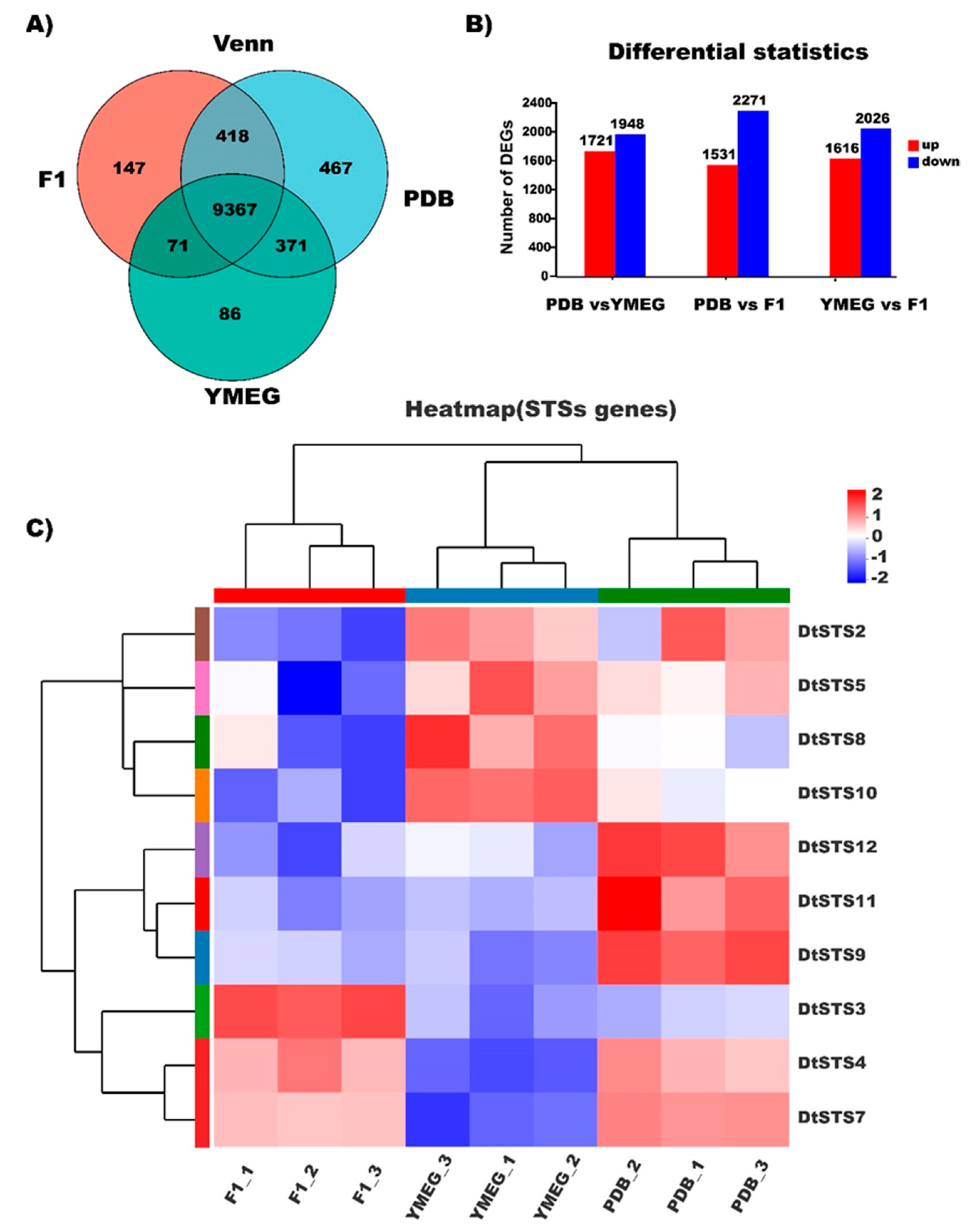
3.5. RNA-Sequencing Based Transcriptome Analysis of the Strain CPCC 401429
3.6. Heterologous Expression of STSs Encoding Genes in Yeast Produced Diverse Sesquiterpenes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christianson, D.W. Unearthing the roots of the terpenome. Curr. Opin. Chem. Biol. 2008, 12, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, X.; Duan, Y.; Wang, P.; Qi, J.; Gao, J.; Liu, C. Biosynthesis of sesquiterpenes in basidiomycetes: A review. J. Fungi 2022, 8, 913. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, J.; Feng, T.; Yang, H.; Ai, H.; Li, Z.; Liu, J. Antroalbocin A, an antibacterial sesquiterpenoid from higher fungus Antrodiella albocinnamomea. Org. Lett. 2018, 20, 8019–8021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cai, G.; Rong, X.; Wang, Y.; Gong, K.; Liu, W.; Wang, L.; Pang, X.; Yu, L. A combination of genome mining with an OSMAC approach facilitates the discovery of and ccontributions to the biosynthesis of melleolides from the basidiomycete Armillaria tabescens. J. Agric. Food Chem. 2022, 70, 12430–12441. [Google Scholar] [CrossRef]
- Lehmann, V.K.; Huang, A.; Ibanez-Calero, S.; Wilson, G.R.; Rinehart, K.L. Illudin S, the sole antiviral compound in mature fruiting bodies of Omphalotus illudens. J. Nat. Prod. 2003, 66, 1257–1258. [Google Scholar] [CrossRef]
- Yang, X.Y.; Feng, T.; Li, Z.; Sheng, Y.; Yin, X.; Leng, Y.; Liu, J. Conosilane A, an unprecedented sesquiterpene from the cultures of basidiomycete Conocybe siliginea. Org. Lett. 2012, 14, 5382–5384. [Google Scholar] [CrossRef]
- Qi, Q.Y.; Bao, L.; Ren, J.; Han, J.; Zhang, Z.; Li, Y.; Yao, Y.; Cao, R.; Liu, H. Sterhirsutins A and B, two new heterodimeric sesquiterpenes with a new skeleton from the culture of Stereum hirsutum collected in Tibet Plateau. Org. Lett. 2014, 16, 5092–5095. [Google Scholar] [CrossRef]
- Agger, S.; Lopez-Gallego, F.; Schmidt-Dannert, C. Diversity of sesquiterpene synthases in the basidiomycete Coprinus cinereus. Mol. Microbiol. 2009, 72, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Wawrzyn, G.T.; Quin, M.B.; Choudhary, S.; Lopez-Gallego, F.; Schmidt-Dannert, C. Draft genome of Omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in Basidiomycota. Chem. Biol. 2012, 19, 772–783. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, X.; Orban, A.; Shukal, S.; Birk, F.; Too, H.P.; Ruhl, M. Agrocybe aegerita serves as a gateway for identifying sesquiterpene biosynthetic enzymes in higher fungi. ACS Chem. Biol. 2020, 15, 1268–1277. [Google Scholar] [CrossRef]
- Yap, H.Y.; Muria-Gonzalez, M.J.; Kong, B.H.; Stubbs, K.A.; Tan, C.S.; Ng, S.T.; Tan, N.H.; Solomon, P.S.; Fung, S.Y.; Chooi, Y.H. Heterologous expression of cytotoxic sesquiterpenoids from the medicinal mushroom Lignosus rhinocerotis in yeast. Microb. Cell Factories 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Onsando, J.M.; Wargo, P.M.; Waudo, S.W. Distribution, severity, and spread of Armillaria root disease in Kenya tea plantations. Plant Dis. 1997, 81, 133–137. [Google Scholar] [CrossRef]
- Baumgartner, K.; Coetzee, M.P.; Hoffmeister, D. Secrets of the subterranean pathosystem of Armillaria. Mol. Plant Pathol. 2011, 12, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Fradj, N.; de Montigny, N.; Merindolm, N.; Awwad, F.; Boumghar, Y.; Germain, H.; Desgagne-Penix, I. A first insight into North American plant pathogenic fungi Armillaria sinapina transcriptome. Biology 2020, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Wang, H.C.; Xue, W.Q.; Zhao, J.; Yang, Z.L. Phylogenetic analyses of Armillaria reveal at least 15 phylogenetic lineages in China, seven of which are associated with cultivated Gastrodia elata. PLoS ONE 2016, 11, e0154794. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.; Travadon, R.; Bruhn, J.; Bergemann, S.E. Contrasting patterns of genetic diversity and population structure of Armillaria mellea sensu stricto in the eastern and western United States. Phytopathology 2010, 100, 708–718. [Google Scholar] [CrossRef]
- Muszynska, B.; Sulkowska-Ziaja, K.; Wolkowska, M.; Ekiert, H. Chemical, pharmacological, and biological characterization of the culinary-medicinal honey mushroom, Armillaria mellea (Vahl) P. Kumm. (Agaricomycetideae): A review. Int. J. Med. Mushrooms 2011, 13, 167–175. [Google Scholar] [CrossRef]
- Hobbs, C. Medicinal Mushrooms: An Exploration of Tradition, Healing, and Culture; Botanica Press: Summertown, TN, USA, 1986. [Google Scholar]
- Zhang, T.; Du, Y.; Liu, X.; Sun, X.; Cai, E.; Zhu, H.; Zhao, Y. Study on antidepressant-like effect of protoilludane sesquiterpenoid aromatic esters from Armillaria mellea. Nat. Prod. Res. 2021, 35, 1042–1045. [Google Scholar] [CrossRef]
- Heinzelmann, R.; Rigling, D.; Sipos, G.; Munsterkotter, M.; Croll, D. Chromosomal assembly and analyses of genome-wide recombination rates in the forest pathogenic fungus Armillaria ostoyae. Heredity 2020, 124, 699–713. [Google Scholar] [CrossRef]
- Akulova, V.S.; Sharov, V.V.; Aksyonova, A.I.; Putintseva, Y.A.; Oreshkova, N.V.; Feranchuk, S.I.; Kuzmin, D.A.; Pavlov, I.N.; Litovka, Y.A.; Krutovsky, K.V. De novo sequencing, assembly and functional annotation of Armillaria borealis genome. BMC Genom. 2020, 21, 534. [Google Scholar] [CrossRef]
- Zhan, M.; Tian, M.; Wang, W.; Li, G.; Lu, X.; Cai, G.; Yang, H.; Du, G.; Huang, L. Draft genomic sequence of Armillaria gallica 012m: Insights into its symbiotic relationship with Gastrodia elata. Braz. J. Microbiol. 2020, 51, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Sipos, G.; Prasanna, A.N.; Walter, M.C.; O’Connor, E.; Balint, B.; Krizsan, K.; Kiss, B.; Hess, J.; Varga, T.; Slot, J.; et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 2017, 1, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Keane, T.M.; Turner, D.J.; O’Keeffe, G.; Fitzpatrick, D.A.; Doyle, S. Genomic and proteomic dissection of the ubiquitous plant pathogen, Armillaria mellea: Toward a new infection model system. J. Proteome Res. 2013, 12, 2552–2570. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.A.; Wilson, A.W.; Sene, O.; Henkel, T.W.; Aime, M.C. Resolved phylogeny and biogeography of the root pathogen Armillaria and its gasteroid relative, Guyanagaster. BMC Evol. Biol. 2017, 17, 33. [Google Scholar] [CrossRef]
- Liang, J.; Pecoraro, L.; Cai, L.; Yuan, Z.; Zhao, P.; Tsui, C.K.M.; Zhang, Z. Phylogenetic relationships, speciation, and origin of Armillaria in the Northern hemisphere: A lesson based on rRNA and elongation factor 1-alpha. J. Fungi 2021, 7, 1088. [Google Scholar] [CrossRef]
- Watling, R.; Kile, G.A.; Gregory, N.M. The genus Armillaria-nomenclature, typification, the identity of Armillaria mellea and species differentiation. Trans. Br. Mycol. Soc. 1982, 78, 271–285. [Google Scholar] [CrossRef]
- Zhang, T.; Gu, G.; Liu, G.; Su, J.; Zhan, Z.; Zhao, J.; Qian, J.; Cai, G.; Cen, C.; Zhang, D.; et al. Late-stage cascade of oxidation reactions during the biosynthesis of oxalicine B in Penicillium oxalicum. Acta Pharm. Sin. B 2023, 13, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Pang, X.; Zhao, J.; Guo, Z.; He, W.; Cai, G.; Su, J.; Cen, S.; Yu, L. Discovery and activation of the cryptic cluster from Aspergillus sp. CPCC 400735 for asperphenalenone biosynthesis. ACS Chem. Biol. 2022, 17, 1524–1533. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M. Usadel B: Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Nguyen, T.; Tagett, R.; Diaz, D.; Draghici, S. A novel approach for data integration and disease subtyping. Genome Res. 2017, 27, 2025–2039. [Google Scholar] [CrossRef]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Peitsch, M.C.; Wilkins, M.R.; Tonella, L.; Sanchez, J.C.; Appel, R.D.; Hochstrasser, D.F. Large-scale protein modelling and integration with the SWISS-PROT and SWISS-2DPAGE databases: The example of Escherichia coli. Electrophoresis 1997, 18, 498–501. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramirez, F.; Vesztrocy, A.W.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.R.; Quijada, L.; Smith, C.A.; Baral, H.O.; Hosoya, T.; Baschien, C.; Partel, K.; Zhuang, W.Y.; Haelewaters, D.; Park, D.; et al. A multigene phylogeny toward a new phylogenetic classification of Leotiomycetes. IMA Fungus 2019, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Koch, R.A.; Herr, J.R. Global distribution and richness of Armillaria and related species inferred from public databases and amplicon sequencing datasets. Front. Microbiol. 2021, 12, 733159. [Google Scholar] [CrossRef]
- Engels, B.; Heinig, U.; Grothe, T.; Stadler, M.; Jennewein, S. Cloning and characterization of an Armillaria gallica cDNA encoding protoilludene synthase, which catalyzes the first committed step in the synthesis of antimicrobial melleolides. J. Biol. Chem. 2011, 286, 6871–6878. [Google Scholar] [CrossRef]
- Raffaele, S.; Kamoun, S. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat. Rev. Microbiol. 2012, 10, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, H.; Zhang, X.; Chen, Q. The genomic and transcriptomic analyses of Floccularia luteovirens, a rare edible fungus in the Qinghai-Tibet Plateau, provide insights into the taxonomy placement and fruiting body formation. J. Fungi 2021, 7, 887. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Jia, Q.; Mu, X.; Hu, B.; Sun, X.; Deng, Z.; Chen, F.; Bian, G.; Liu, T. Systematic mining of fungal chimeric terpene synthases using an efficient precursor-providing yeast chassis. Proc. Natl. Acad. Sci. USA 2021, 118, e2023247118. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Description | Source |
|---|---|---|
| D. tabescens CPCC 401429 | Melleolides producing strain | [4] |
| E. coli Trans T1 | F-φ80 lac ZΔM15 Δ(lacZYA-arg F) U169 endA1 recA1 hsdR17(rk-,mk+) supE44λ- thi -1 gyrA96 relA1 phoA | Transgen |
| S. cerevisiae BJ5464 | (MATα ura3-52 his3-Δ200 leu2- Δ1 trp1 pep4::HIS3 prb1 Δ1.6R can1 GAL | [29] |
| BJ-CK | S. cerevisiae BJ 5464, carrying plasmid YET | This study |
| BJ-DtSTS9 | S. cerevisiae BJ 5464, carrying plasmid YET-DtSTS9 | This study |
| BJ-DtSTS10 | S. cerevisiae BJ 5464, carrying plasmid YET-DtSTS10 | This study |
| Plasmids | Description | Source |
| YET | tryptophan auxotrophic expressing plasmid of yeast | [29] |
| YET-DtSTS9 | YET expressing plasmid with gene DtSTS9 | This study |
| YET-DtSTS10 | YET expressing plasmid with gene DtSTS10 | This study |
| Primers | Sequence | Size (bp) |
| DtSTS9-F | atacaatcaactatcaactattaactatatcgtaataccatatgacactatctacagca | 963 bp |
| DtSTS9-R | cttgataatggaaactataaatcgtgaaggcatgtttaaacttataatcccagctcgct | |
| DtSTS10-F | atacaatcaactatcaactattaactatatcgtaataccatatgactttatctacttcg | 1005 bp |
| DtSTS10-R | cttgataatggaaactataaatcgtgaaggcatgtttaaacctatatgccaagctcgct |
| Genome Feature | No./Value |
|---|---|
| Genome size (Mb) | 50.36 |
| Number of scaffolds | 713 |
| Scaffold N50 (bp) | 125,925 |
| Scaffold N90 (bp) | 33,591 |
| Longest scaffold (bp) | 839,203 |
| Shortest scaffold (bp) | 526 |
| GC content (%) | 47.14% |
| Protein-coding genes | 15,145 |
| Average gene length (bp) | 1959 |
| Complete BUSCOs (%) | 92.4% |
| Genes of KEGG | 3441 |
| Genes of COG | 9507 |
| tRNA No. | 255 |
| rRNA No. | 4 |
| TE No. | 438 |
| Rt (min) | Peak Area (%) | Name/Structure | Rt (min) | Peak Area (%) | Name/Structure |
|---|---|---|---|---|---|
| BJ-At_STS9 | BJ-At_STS10 | ||||
| 33.563 | 2.61 |  α-bulnesene | 35.938 | 1.86 |  cis-thujopsene |
| 34.167 | 1.29 |  (+)-aromadendrene | 38.618 | 0.54 |  acoradiene |
| 34.866 | 3.6 |  4,10-dimethyl-7-isopropyl [4,4,0]-bicyclo-1,4-decadiene | 38.87 | 1.55 |  α-cedrene |
| 35.971 | 11.25 |  cis-thujopsene | 39.39 | 0.88 |  γ-muurolene |
| 38.627 | 3.68 |  acoradiene | 39.959 | 0.37 |  δ-elemene |
| 39.699 | 4.4 |  α-cubebene | 41.567 | 83.59 |  α-himachalene |
| 40.243 | 1.66 |  β-chamigrene | 41.924 | 2.0 |  β-himachalene |
| 41.511 | 0.68 |  thujopsene-(I2) | 43.573 | 2.9 |  β-sesquiphellandrene |
| 41.917 | 4.82 |  β-himachalene | 45.864 | 2.0 |  (+)-cuparene |
| 45.865 | 1.41 |  (+)-cuparene | 54.483 | 4.62 |  α-bisabolol |
| 53.606 | 2.14 |  widdrol | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Feng, J.; He, W.; Rong, X.; Lv, H.; Li, J.; Li, X.; Wang, H.; Wang, L.; Zhang, L.; et al. Genomic and Transcriptomic Approaches Provide a Predictive Framework for Sesquiterpenes Biosynthesis in Desarmillaria tabescens CPCC 401429. J. Fungi 2023, 9, 481. https://doi.org/10.3390/jof9040481
Zhang T, Feng J, He W, Rong X, Lv H, Li J, Li X, Wang H, Wang L, Zhang L, et al. Genomic and Transcriptomic Approaches Provide a Predictive Framework for Sesquiterpenes Biosynthesis in Desarmillaria tabescens CPCC 401429. Journal of Fungi. 2023; 9(4):481. https://doi.org/10.3390/jof9040481
Chicago/Turabian StyleZhang, Tao, Jianjv Feng, Wenni He, Xiaoting Rong, Hui Lv, Jun Li, Xinxin Li, Hao Wang, Lu Wang, Lixin Zhang, and et al. 2023. "Genomic and Transcriptomic Approaches Provide a Predictive Framework for Sesquiterpenes Biosynthesis in Desarmillaria tabescens CPCC 401429" Journal of Fungi 9, no. 4: 481. https://doi.org/10.3390/jof9040481
APA StyleZhang, T., Feng, J., He, W., Rong, X., Lv, H., Li, J., Li, X., Wang, H., Wang, L., Zhang, L., & Yu, L. (2023). Genomic and Transcriptomic Approaches Provide a Predictive Framework for Sesquiterpenes Biosynthesis in Desarmillaria tabescens CPCC 401429. Journal of Fungi, 9(4), 481. https://doi.org/10.3390/jof9040481