
Genomic and Transcriptomic Survey Provides Insights into Molecular Basis of Pathogenicity of the Sunflower Pathogen Phoma macdonaldii

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Growth Conditions
2.2. Phylogenetic Analysis
2.3. Genome Sequencing and Assembly
2.4. Genomic Prediction and Genome Annotation
2.5. Pathogenicity Assay
2.6. mRNA Library Constructing and Sequencing
2.7. Data Mining and Differential Expression Analysis
2.8. RNA Preparation and Quantitative Real-Time (qRT)-PCR
3. Results
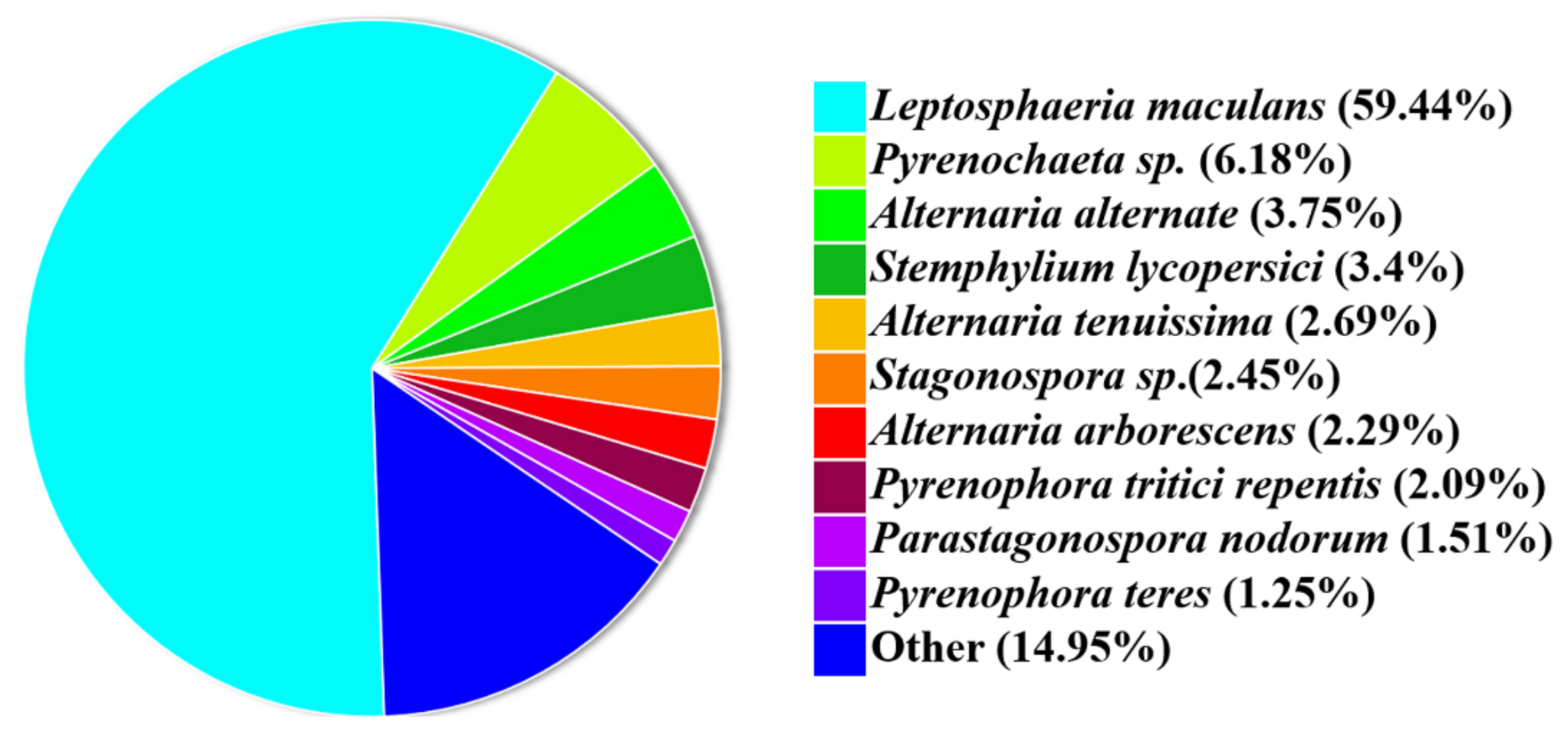
3.1. Phylogenic Analysis of P. macdonaldii CXJ0811
3.2. Genome Structures
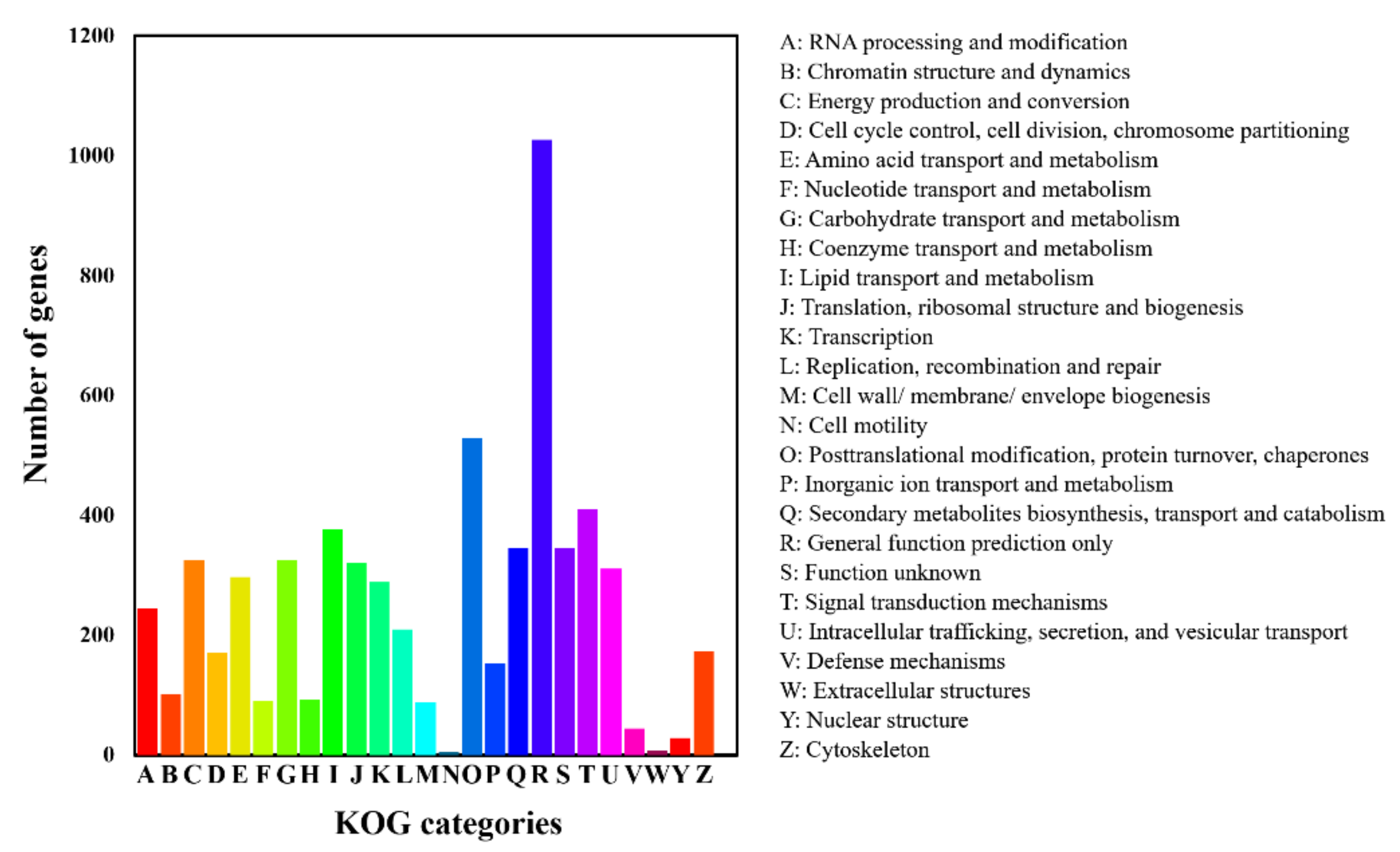
3.3. Gene Annotation
3.4. Genes Involved in Carbohydrate Degradation (CAZymes)
3.5. Pathogenesis Related Genes
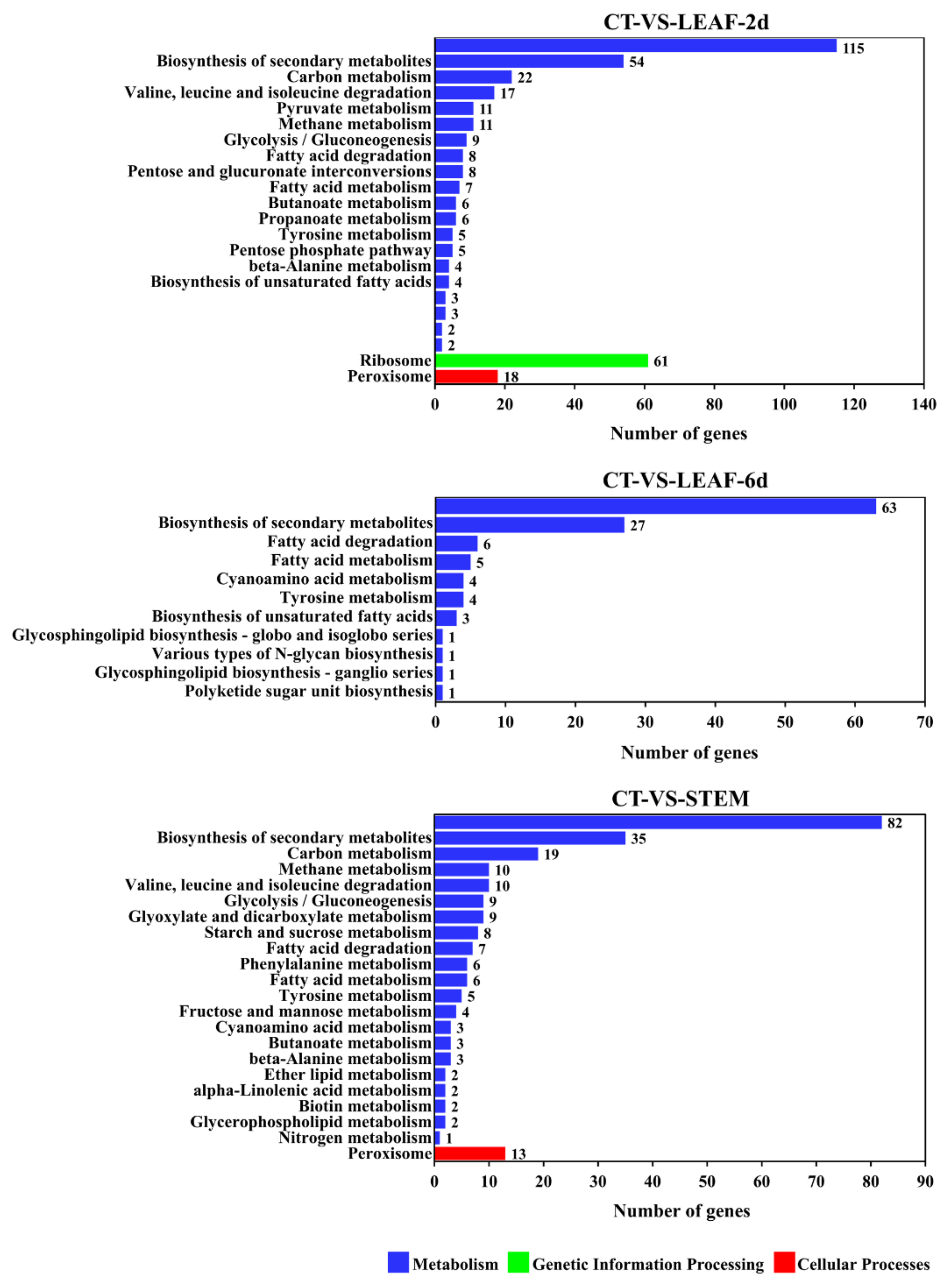
3.6. Transcriptomic Pattern of Genes from Different Diseased Tissues
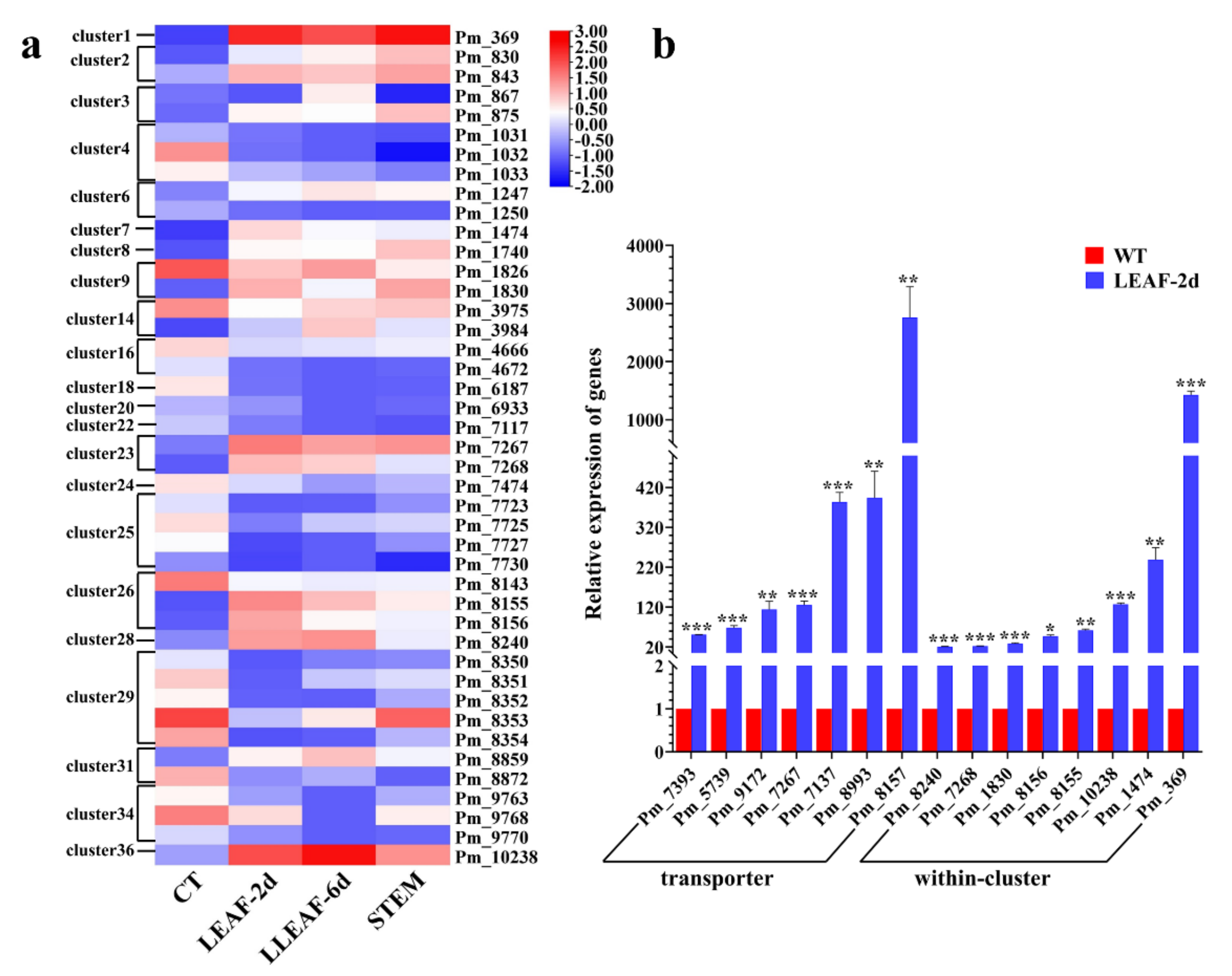
3.7. Transcriptomic Pattern of Pathogenesis Genes
4. Discussion
4.1. Role of Plant Cell Wall-Degrading Enzymes (CWDEs) in Pathogenesis of P. macdonaldii
4.2. Role of Effectors in Pathogenesis of P. macdonaldii
4.3. Role of Phytotoxins in Pathogenesis of P. macdonaldii
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAOSTA. Food and Agriculture Data. Available online: http://www.fao.org/faostat/ (accessed on 20 October 2022).
- Masirevic, S. Sunflower diseases research progress and management. In Proceedings of the 19th International Sunflower Con-ference, Edirne, Turkey, 29 May–3 June 2016. [Google Scholar]
- Derevnina, L.; Petre, B.; Kellner, R.; Dagdas, Y.F.; Sarowar, M.N.; Giannakopoulou, A. Emerging oomycete threats to plants and animals. Philos. Trans. R. Soc. B: Biol. Sci. 2016, 371, 20150459. [Google Scholar] [CrossRef]
- Miric, E.; Aitken, E.A.B.; Goulter, K.C. Identification in Australia of the quarantine pathogen of sunflower Phoma macdonaldii (Teleomorph: Leptosphaeria lindquistii). Aust. J. Agric. Res. 1999, 50, 325–332. [Google Scholar] [CrossRef]
- Debaekea, P.; Pérès, A. Influence of sunflower (Helianthus annuus L.) crop management on Phoma black stem (Phoma macdonaldii Boerema). Crop Prot. 2003, 22, 741–752. [Google Scholar] [CrossRef]
- Seassau, C.; Debaeke, P.; Mestries, E.; Dechamp-Guillaume, G. Evaluation of Inoculation Methods to Reproduce Sunflower Premature Ripening Caused by Phoma macdonaldii. Plant Dis. 2010, 94, 1398–1404. [Google Scholar] [CrossRef]
- McDonald, W.C. Phoma black stem of sunflowers. Phytopathology 1964, 54, 492–493. [Google Scholar]
- Adeleke, B.S.; Babalola, O.O. Oilseed crop sunflower (Helianthus annuus) as a source of food: Nutritional and health benefits. Food Sci. Nutr. 2020, 8, 4666–4684. [Google Scholar] [CrossRef]
- Liu, L.; Li, X. The geographical distribution of sunflower diseases in China. Plant Pathology 1988, 37, 470–474. [Google Scholar] [CrossRef]
- Chen, W.M.; Guo, Q.Y.; Song, H.M.; Wang, H.; Ma, F.J.; Jing, X.Y. Domestic new plant disease: The first report of the occurrence of sunflower Phoma black stem in Ili River Valley, Sinkiang, China. J. Yunnan Agric. Univ. 2008, 23, 609–612. [Google Scholar]
- Yan, N.N.; Na, R.; Jia, R.F.; Zhang, J.; Zhao, J. Occurrence of black stem on Helianthus annuus caused by Phoma macdonaldii and resistant evaluation of different sunflower varieties. Oilseeds Fats Crops Lipids 2020, 27, 1–5. [Google Scholar] [CrossRef]
- Bordat, A.; Marchand, G.; Langlade, N.B.; Pouilly, N.; Muños, S.; Dechamp-Guillaume, G.; Vincourt, P.; Bret-Mestries, E. Different genetic architectures underlie crop responses to the same pathogen: The {Helianthus annuus * Phoma macdonaldii} interaction case for black stem disease and premature ripening. BMC Plant Biol. 2017, 17, 167. [Google Scholar] [CrossRef]
- Al-Chaarani, G.R.; Roustaee, A.; Gentzbittel, L.; Mokrani, L.; Barrault, G.; Dechamp-Guillaume, G.; Sarrafi, A. A QTL analysis of sunflower partial resistance to downy mildew (Plasmopara halstedii) and black stem (Phoma macdonaldii) by the use of re-combinant inbred lines (RILs). Theor. Appl. Genet. 2002, 104, 490–496. [Google Scholar] [CrossRef]
- Alignan, M.; Hewezi, T.; Petitprez, M.; Dechamp-Guillaume, G.; Gentzbittel, L. A cDNA microarray approach to decipher sunflower (Helianthus annuus) responses to the necrotrophic fungus Phoma macdonaldii. New Phytol. 2006, 170, 523–536. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Yu, Y. Phytotoxins, elicitors and other secondary metabolites from phytopathogenic “blackleg” fungi: Structure, phytotoxicity and biosynthesis. Nat. Prod. Commun. 2009, 4, 1291–1304. [Google Scholar] [CrossRef]
- Chen, Q.; Jiang, J.R.; Zhang, G.Z.; Cai, L.; Crous, P.W. Resolving the Phoma enigma. Stud. Mycol. 2015, 82, 137–217. [Google Scholar] [CrossRef]
- Roustaee, A.; Costes, S.; Dechamp-Guillaume, G.; Barrault, G. Phenotypic variability of Leptosphaeria lindquistii (anamorph: Phoma macdonaldii), a fungal pathogen of sunflower. Plant Pathol. 2000, 49, 227–234. [Google Scholar] [CrossRef]
- Sun, H.; Kav, N.N.V.; Liang, Y.; Sun, L.; Chen, W. Proteome of the fungus Phoma macdonaldii, the causal agent of black stem of sunflower. J. Proteom. 2020, 225, 103878. [Google Scholar] [CrossRef]
- McCarthy, A. Third Generation DNA Sequencing: Pacific Biosciences’ Single Molecule Real Time Technology. Chem. Biol. 2010, 17, 675–676. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. J. Genet. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Mario, S.; Rasmus, S.; Stephan, W.; Burkhard, M. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, 309–312. [Google Scholar]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Microbial genome analysis: The COG approach. Briefings Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 4, D353–D361. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Im-proving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 2, W29–W35. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database J. Biol. Databases Curation 2012, 22, bas032. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 1, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ait-Lahsen, H.; Soler, A.; Rey, M.; de La Cruz, J.; Monte, E.; Llobell, A. An antifungal exo-alpha-1, 3-glucanase (AGN13.1) from The biocontrol fungus Trichoderma harzianum. Appl. Environ. Microbiol. 2001, 67, 5833–5839. [Google Scholar] [CrossRef]
- Scherlach, K.; Boettger, D.; Remme, N.; Hertweck, C. The chemistry and biology of cytochalasans. Nat. Prod. Rep. 2010, 27, 869–886. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Nair, R.; Banerjee, A. Multidrug transporters of Candida species in clinical azole resistance. Fungal Genet. Biol. 2019, 132, 103252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Correction: Comparative analysis of fungal genomes reveals different plant cell wall de-grading capacity in fungi. BMC Genom. 2014, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Senthilraja, G.; Anand, T.; Mohankumar, S.; Raguchander, T.; Samiyappan, R. Analysis of variation in virulence of Beauveria bassiana against insect pests of pigeonpea using qPCR. J. Basic Microbiol. 2018, 58, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Minio, A.; Ferrarini, A.; Valente, M.T.; Bagnaresi, P.; Orrù, L.; Tononi, P.; Zamperin, G.; Infantino, A.; Valè, G.; et al. De novo genome assembly of the soil-borne fungus and tomato pathogen Pyrenochaeta lycopersici. BMC Genom. 2014, 15, 313. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Yao, W.; Duan, Z.; Powell, C.A.; Chen, B.; Zhang, M. Genome Sequence of Phoma sorghina var. saccharum That Causes Sugarcane Twisted Leaf Disease in China. Mol. Plant-Microbe Interact. 2020, 33, 1092–1094. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, M.; Wu, J.; Dong, W.; Chen, D.; Wang, L.; Chi, Y. Draft Genome Sequence of Phoma arachidicola Wb2 Causing Peanut Web Blotch in China. Curr. Microbiol. 2019, 76, 200–206. [Google Scholar] [CrossRef]
- Matar, K.A.O.; Chen, X.; Chen, D.; Anjago, W.M.; Norvienyeku, J.; Lin, Y.; Chen, M.; Wang, Z.; Ebbole, D.J.; Lu, G.-D. WD40-repeat protein MoCreC is essential for carbon repression and is involved in conidiation, growth and pathogenicity of Magnaporthe oryzae. Curr. Genet. 2017, 63, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Taschuk, F.; Cherry, S. DEAD-Box Helicases: Sensors, Regulators, and Effectors for Antiviral Defense. Viruses 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, T.J.; Dunkle, L.D.; Ciuffetti, L.M. Host-selective toxins and avirulence determinants: What’s in a name? Annu. Rev. Phytopathol. 2002, 40, 251–285. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Su, Y.; Zhou, S.; Feng, Y.; Guo, W.; Wang, X. A RACK1-like protein regulates hyphal morphogenesis, root entry and in vivo virulence in Verticillium dahliae. Fungal Genet. Biol. 2017, 99, 52–61. [Google Scholar] [CrossRef]
- Molinero-Ruiz, L. Sustainable and efficient control of sunflower downy mildew by means of genetic resistance: A review. Theor. Appl. Genet. 2022, 135, 3757–3771. [Google Scholar] [CrossRef]
- Darvishzadeh, R.; Hewezi, T.; Gentzbittel, L.; Sarrafi, A. Differential expression of defence-related genes between compatible and partially compatible sunflower–Phoma macdonaldii interactions. Crop. Prot. 2008, 27, 740–746. [Google Scholar] [CrossRef]
- Isaac, S. Fungal-Plant Interactions; Chapman and Hall: London, UK, 1992. [Google Scholar]
- Roustaee, A.; Dechamp-Guillaume, G.; Gelie, B.; Savy, C.; Dargent, R.; Barrault, G. Ultrastructural Studies of the Mode of Penetration by Phoma macdonaldii in Sunflower Seedlings. Phytopathology® 2000, 90, 915–920. [Google Scholar] [CrossRef]
- Doehlemann, G.; Ökmen, B.; Zhu, W.; Sharon, A. Plant pathogenic fungi. Microbiology spectrum. 2017, 5. [Google Scholar]
- Zhang, L.; Kars, I.; Essenstam, B.; Liebrand, T.W.H.; Wagemakers, L.; Elberse, J.; Tagkalaki, P.; Tjoitang, D.; van den Ackerveken, G.; van Kan, J.A.L. Fungal Endopolygalacturonases Are Recognized as Microbe-Associated Molecular Patterns by the Arabidopsis Receptor-Like Protein RESPONSIVENESS TO BOTRYTIS POLYGALACTURONASES1. Plant Physiol. 2014, 164, 352–364. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Qiu, D.; Zeng, H.; Guo, L.; Yang, X. BcGs1, a glycoprotein from Botrytis cinerea, elicits defence response and improves disease resistance in host plants. Biochem. Biophys. Res. Commun. 2015, 457, 627–634. [Google Scholar] [CrossRef]
- Brito, N.; Espino, J.J.; González, C. The endo-beta-1,4-xylanase xyn11A is required for virulence in Botrytis cinerea. Mol. Plant-Microbe Interact. 2006, 19, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ben-Daniel, B.-H.; Bar-Zvi, D.; Tsror Lahkim, L. Pectate lyase affects pathogenicity in natural isolates of Colletotrichum coccodes and in pelA gene-disrupted and gene-overexpressing mutant lines. Mol. Plant Pathol. 2012, 13, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Abdul Malik, N.A.; Kumar, I.S.; Nadarajah, K. Elicitor and Receptor Molecules: Orchestrators of Plant Defense and Immunity. Int. J. Mol. Sci. 2020, 21, 963. [Google Scholar] [CrossRef]
- Flor, H.H. Current Status of the Gene-For-Gene Concept. Annu. Rev. Phytopathol. 1971, 9, 275–296. [Google Scholar] [CrossRef]
- El Oirdi, M.; El Rahman, T.A.; Rigano, L.; El Hadrami, A.; Rodriguez, M.C.; Daayf, F.; Vojnov, A.; Bouarab, K. Botrytis cinerea Manipulates the Antagonistic Effects between Immune Pathways to Promote Disease Development in Tomato. Plant Cell 2011, 23, 2405–2421. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Xie, J.; Cheng, J.; Li, G.; Yi, X.; Jiang, D.; Fu, Y. Novel secretory protein Ss-Caf1 of the plant-pathogenic fungus Scle-rotinia sclerotiorum is required for host penetration and normal sclerotial development. Mol. Plant-Microbe Interact. 2014, 27, 40–55. [Google Scholar] [CrossRef]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef]
- Lyu, X.; Shen, C.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Cheng, J. A small secreted virulence-related protein is essential for the ne-crotrophic interactions of Sclerotinia sclerotiorum with its host plants. PLoS Pathog. 2016, 12, e1005435. [Google Scholar] [CrossRef]
- Dallal Bashi, Z.; Hegedus, D.D.; Buchwaldt, L.; Rimmer, S.R.; Borhan, M.H. Expression and regulation of Sclerotinia sclerotiorum ne-crosis and ethylene-inducing peptides (NEPs). Mol. Plant Pathol. 2010, 11, 43–53. [Google Scholar] [CrossRef]
- Xu, D.; Xue, M.; Shen, Z.; Jia, X.; Hou, X.; Lai, D.; Zhou, L. Phytotoxic Secondary Metabolites from Fungi. Toxins 2021, 13, 261. [Google Scholar] [CrossRef]
- Parisi, A.; Piattelli, M.; Tringali, C.; Di San Lio, G.M. Indentificaton of the phytotoxin mullein in culture fluids of Phoma tra-cheiphila. Phytochemistry 1993, 32, 865–867. [Google Scholar] [CrossRef]
- Smith, G.R.; Munro, M.H.G.; Fineran, B.A.; Cole, A.L.J. Evidence for the involvement of ascochitine in phoma leafspot-wilt disease of Clematis. Physiol. Mol. Plant Pathol. 1994, 45, 333–348. [Google Scholar] [CrossRef]
- Ichihara, A.; Oikawa, H.; Hayashi, K.; Hashimoto, M.; Sakamura, S.; Sakai, R. 3-Deoxyaphidicolin and Aphidi-colin Analogues as Phytotoxins fromPhoma betae. Agric. Biol. Chem. 1984, 48, 1687–1689. [Google Scholar] [CrossRef]
- Quereshi, S.; Khan, N.A.; Pandey, A.K. Anthraquinone pigment with herbicidal potential from Phoma herbarum FGCC#54. Chem. Nat. Compd. 2011, 47, 521–523. [Google Scholar] [CrossRef]
- Evidente, M.; Cimmino, A.; Zonno, M.C.; Masi, M.; Berestetskyi, A.; Santoro, E.; Superchi, S.; Vurro, M.; Evidente, A. Phytotoxins produced by Phoma chenopodiicola, a fungal pathogen of Chenopodium album. Phytochemistry 2015, 117, 482–488. [Google Scholar] [CrossRef]
- Evidente, A.; Lanzetta, R.; Capasso, R.; Andolfi, A.; Vurro, M.; Zonno, M.C. Putaminoxins B and C from Phoma putaminum. Phytochemistry 1997, 44, 1041–1045. [Google Scholar] [CrossRef]
- Evidente, A.; Randazzo, G.; Iacobellis, N.S.; Bottalico, A. Structure of Cavoxin, a New Phytotoxin from Phoma cava and Cavoxone, Its Related Chroman-4-One. J. Nat. Prod. 1985, 48, 916–923. [Google Scholar] [CrossRef]
- Ichihara, A.; Sawamura, S.; Kawakami, Y.; Sakamura, S. Dihydrogladiolic acid, another phytotoxin from Phoma asparagi Sacc. Agric. Biol. Chem. 1985, 49, 1891–1892. [Google Scholar]
- Capasso, R.; Iacobellis, N.S.; Bottalico, A.; Randazzo, G. Structure-toxicity relationships of the eremophilane phomenone and PR-toxin. Phytochemistry 1984, 23, 2781–2784. [Google Scholar] [CrossRef]
- Cimmino, A.; Andolfi, A.; Berestetskiy, A.; Evidente, A. Production of Phytotoxins by Phoma exigua var. exigua, a Potential Mycoherbicide against Perennial Thistles. J. Agric. Food Chem. 2008, 56, 6304–6309. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Value |
|---|---|
| Number of contigs | 27 |
| Total contigs size (bp) | 38,245,005 |
| Min contigs length (bp) | 40,900 |
| Max contigs length (bp) | 2,753,394 |
| N50 contigs (bp) | 1,508,676 |
| Contig L50 | 10 |
| GC content contigs (%) | 48.27 |
| Attribute | Value |
|---|---|
| Total genes | 11,094 |
| Protein-coding genes | 10,933 |
| With a KOG annotation | 5564 |
| With a GO annotation | 5696 |
| With a KEGG annotation | 2224 |
| With a Swiss-Prot annotation | 7027 |
| With a Pfam annotation | 7598 |
| With a NR annotation | 10,205 |
| With a CAZy number | 1133 |
| With a PHI number | 2356 |
| With a DFVF number | 2167 |
| Secondary metabolite biosynthetic gene clusters | 37 |
| Genes encoded with secreted proteins | 827 |
| Genes with signal peptides | 1057 |
| Genes with transmembrane helices | 2181 |
| Total size (bp) | 17,974,472 |
| Average gene length (bp) | 1644 |
| Genome coding (%) | 47% |
| Total number of ncRNA | 161 |
| Number of rRNAs | 45 |
| Number of tRNAs | 73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Hao, X.; Akhberdi, O.; Zhu, X. Genomic and Transcriptomic Survey Provides Insights into Molecular Basis of Pathogenicity of the Sunflower Pathogen Phoma macdonaldii. J. Fungi 2023, 9, 520. https://doi.org/10.3390/jof9050520
Chen X, Hao X, Akhberdi O, Zhu X. Genomic and Transcriptomic Survey Provides Insights into Molecular Basis of Pathogenicity of the Sunflower Pathogen Phoma macdonaldii. Journal of Fungi. 2023; 9(5):520. https://doi.org/10.3390/jof9050520
Chicago/Turabian StyleChen, Xuejing, Xiaoran Hao, Oren Akhberdi, and Xudong Zhu. 2023. "Genomic and Transcriptomic Survey Provides Insights into Molecular Basis of Pathogenicity of the Sunflower Pathogen Phoma macdonaldii" Journal of Fungi 9, no. 5: 520. https://doi.org/10.3390/jof9050520
APA StyleChen, X., Hao, X., Akhberdi, O., & Zhu, X. (2023). Genomic and Transcriptomic Survey Provides Insights into Molecular Basis of Pathogenicity of the Sunflower Pathogen Phoma macdonaldii. Journal of Fungi, 9(5), 520. https://doi.org/10.3390/jof9050520