
A Custom Regional DNA Barcode Reference Library for Lichen-Forming Fungi of the Intermountain West, USA, Increases Successful Specimen Identification

Abstract
:1. Introduction
2. Materials and Methods
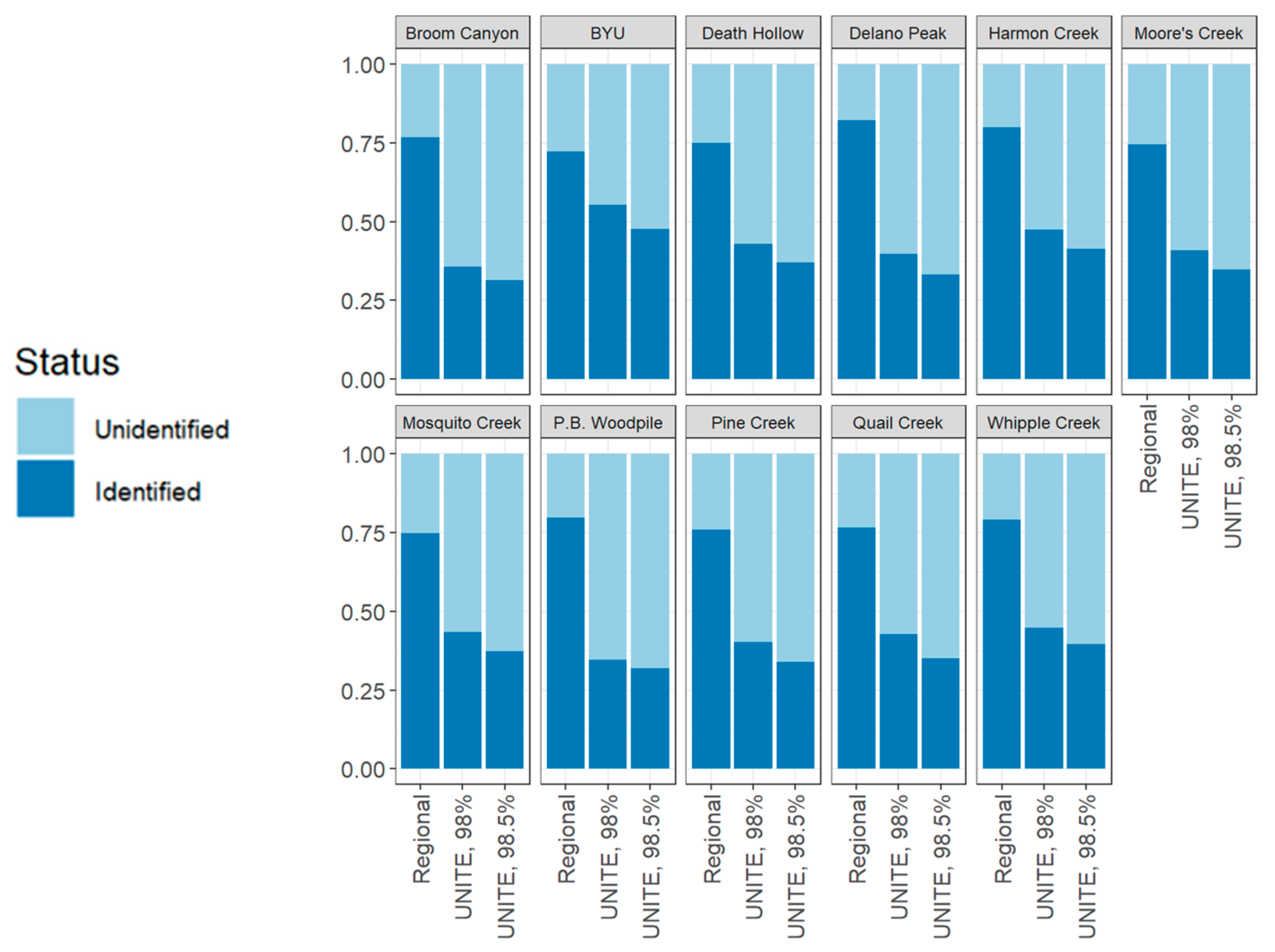
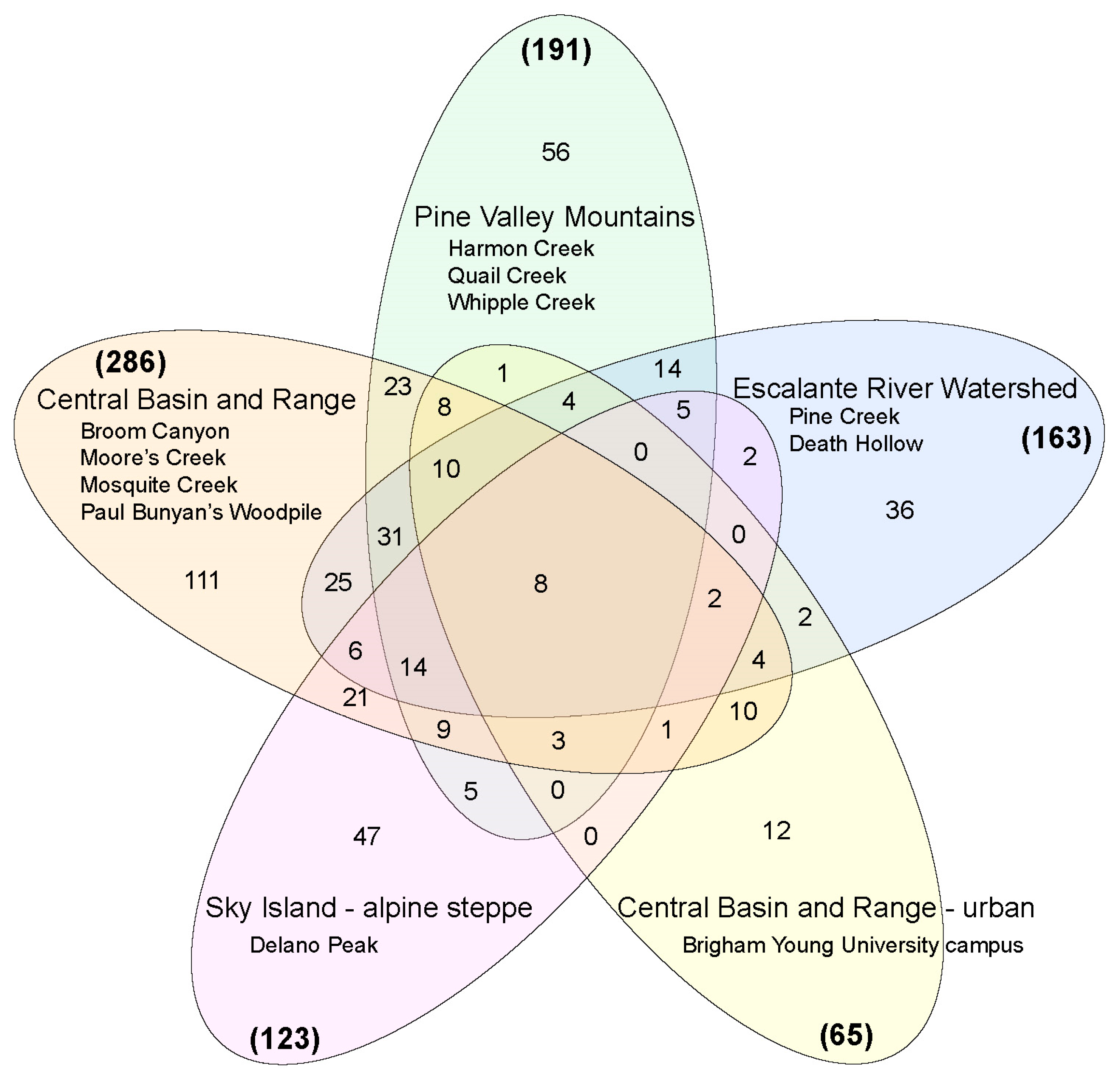
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; McGuire, J.A.; Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 2012, 9, 480–488. [Google Scholar] [CrossRef]
- Naciri, Y.; Linder, H.P. Species delimitation and relationships: The dance of the seven veils. TAXON 2015, 64, 3–16. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Brochmann, C.; Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Zinger, L.; Nilsson, R.H.; Kennedy, P.G.; Yang, T.; Anslan, S.; Mikryukov, V. Best practices in metabarcoding of fungi: From experimental design to results. Mol. Ecol. 2022, 31, 2769–2795. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List; Bolchacova, E.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Phukhamsakda, C.; Nilsson, R.H.; Bhunjun, C.S.; de Farias, A.R.G.; Sun, Y.-R.; Wijesinghe, S.N.; Raza, M.; Bao, D.-F.; Lu, L.; Tibpromma, S.; et al. The numbers of fungi: Contributions from traditional taxonomic studies and challenges of metabarcoding. Fungal Divers. 2022, 114, 327–386. [Google Scholar] [CrossRef]
- Bernard, M.; Rué, O.; Mariadassou, M.; Pascal, G. FROGS: A powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Brief. Bioinform. 2021, 22, bbab318. [Google Scholar] [CrossRef]
- Větrovský, T.; Morais, D.; Kohout, P.; Lepinay, C.; Algora, C.; Awokunle Hollá, S.; Bahnmann, B.D.; Bílohnědá, K.; Brabcová, V.; D’Alò, F.; et al. GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci. Data 2020, 7, 228. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi; recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Baldrian, P.; Větrovský, T.; Lepinay, C.; Kohout, P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers. 2022, 114, 539–547. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 79–95. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Kress, W.J. Plant DNA barcodes: Applications today and in the future. J. Syst. Evol. 2017, 55, 291–307. [Google Scholar] [CrossRef] [Green Version]
- Mohammed-Geba, K.; Obuid-Allah, A.H.; El-Shimy, N.A.; Mahbob, M.A.E.-M.; Ali, R.S.; Said, S.M. DNA Barcoding for Scorpion Species from New Valley Governorate in Egypt Reveals Different Degrees of Cryptic Speciation and Species Misnaming. Conservation 2021, 1, 228–240. [Google Scholar] [CrossRef]
- Mugnai, F.; Meglécz, E.; Abbiati, M.; Bavestrello, G.; Bertasi, F.; Bo, M.; Capa, M.; Chenuil, A.; Colangelo, M.A.; De Clerck, O.; et al. Are well-studied marine biodiversity hotspots still blackspots for animal barcoding? Glob. Ecol. Conserv. 2021, 32, e01909. [Google Scholar] [CrossRef]
- Virgilio, M.; Backeljau, T.; Nevado, B.; De Meyer, M. Comparative performances of DNA barcoding across insect orders. BMC Bioinform. 2010, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Ryberg, M.; Kristiansson, E.; Abarenkov, K.; Larsson, K.-H.; Kõljalg, U. Taxonomic Reliability of DNA Sequences in Public Sequence Databases: A Fungal Perspective. PLoS ONE 2006, 1, e59. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Kristiansson, E.; Ryberg, M.; Nogal-Prata, S.; Gómez-Martínez, D.; Stüer-Patowsky, K.; Jansson, T.; Põlme, S.; Ghobad-Nejhad, M.; Corcoll, N.; et al. The curse of the uncultured fungus. MycoKeys 2022, 86, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Gostel, M.R.; Kress, W.J. The Expanding Role of DNA Barcodes: Indispensable Tools for Ecology, Evolution, and Conservation. Diversity 2022, 14, 213. [Google Scholar] [CrossRef]
- Buckner, J.C.; Sanders, R.C.; Faircloth, B.C.; Chakrabarty, P. The critical importance of vouchers in genomics. eLife 2021, 10, e68264. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, I.A.; Baugh, M.; Henrie, J.R.; Hollinger, J.; Crepeau, R.; Leavitt, S.D. Integrative Biodiversity Inventories: Characterizing Lichen-Forming Fungal Diversity in Glen Canyon National Recreation Area Using DNA Barcoding and Vouchered Specimens. West. N. Am. Nat. 2022, 82, 213–233. [Google Scholar] [CrossRef]
- Zhang, Y.; Clancy, J.; Jensen, J.; McMullin, R.T.; Wang, L.; Leavitt, S.D. Providing Scale to a Known Taxonomic Unknown—At Least a 70-Fold Increase in Species Diversity in a Cosmopolitan Nominal Taxon of Lichen-Forming Fungi. J. Fungi 2022, 8, 490. [Google Scholar]
- Leavitt, S.D.; Hollinger, J.; Summerhays, S.; Munger, I.; Allen, J.; Smith, B. Alpine lichen diversity in an isolated sky island in the Colorado Plateau, USA—Insight from an integrative biodiversity inventory. Ecol. Evol. 2021, 11, 11090–11101. [Google Scholar] [CrossRef]
- Wright, B.; St. Clair, L.L.; Leavitt, S.D. Is targeted community DNA metabarcoding suitable for biodiversity inventories of lichen-forming fungi? Ecol. Indic. 2019, 98, 812–820. [Google Scholar] [CrossRef]
- Henrie, J.R.; Thomson, B.M.; Yungfleisch, A.A.; Kerr, M.; Leavitt, S.D. Characterizing Crustose Lichen Communities—DNA Metabarcoding Reveals More than Meets the Eye. Diversity 2022, 14, 766. [Google Scholar] [CrossRef]
- Wise, E.K. Hydroclimatology of the US Intermountain West. Prog. Phys. Geogr. 2012, 36, 458–479. [Google Scholar] [CrossRef]
- McCune, B.; Yang, S.; Jovan, S.; Root, H.T. Climate and epiphytic macrolichen communities in the Four Corners region of the U.S.A. Bryologist 2022, 125, 70–90. [Google Scholar] [CrossRef]
- Lücking, R.; Leavitt, S.D.; Hawksworth, D.L. Species in lichen-forming fungi: Balancing between conceptual and practical considerations, and between phenotype and phylogenomics. Fungal Divers. 2021, 109, 99–154. [Google Scholar] [CrossRef]
- Spribille, T.; Fryday, A.M.; Pérez-Ortega, S.; Svensson, M.; Tønsberg, T.; Ekman, S.; Holien, H.; Resl, P.; Schneider, K.; Stabentheiner, E.; et al. Lichens and associated fungi from Glacier Bay National Park, Alaska. Lichenologist 2020, 52, 61–181. [Google Scholar] [CrossRef]
- Luo, A.; Ling, C.; Ho, S.Y.W.; Zhu, C.-D. Comparison of Methods for Molecular Species Delimitation Across a Range of Speciation Scenarios. Syst. Biol. 2018, 67, 830–846. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [Green Version]
- United States Environmental Protection Agency. Ecoregions. Available online: https://www.epa.gov/eco-research/ecoregions (accessed on 8 May 2023).
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2017, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. PeerJ 2014, 2, e593. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, E.; Navas-Molina, J.A.; Mercier, C.; Xu, Z.Z.; Mahé, F.; He, Y.; Zhou, H.-W.; Rognes, T.; Caporaso, J.G.; Knight, R. Open-Source Sequence Clustering Methods Improve the State Of the Art. mSystems 2016, 1, e00003-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Rozewicki, J.; Yamada, K.D.; Katoh, K. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree version 1.4. 2008. Available online: http://tree.bio.ed.ac.uk/software/Wgtree/ (accessed on 8 May 2023).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Marthinsen, G.; Rui, S.; Timdal, E. OLICH: A reference library of DNA barcodes for Nordic lichens. Biodivers. Data J. 2019, 7, e36252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Questel, J.M.; Hopcroft, R.R.; Dehart, H.M.; Smoot, C.A.; Kosobokova, K.N.; Bucklin, A. Metabarcoding of zooplankton diversity within the Chukchi Borderland, Arctic Ocean: Improved resolution from multi-gene markers and region-specific DNA databases. Mar. Biodivers. 2021, 51, 4. [Google Scholar] [CrossRef]
- Carew, M.E.; Nichols, S.J.; Batovska, J.; St Clair, R.; Murphy, N.P.; Blacket, M.J.; Shackleton, M.E. A DNA barcode database of Australia’s freshwater macroinvertebrate fauna. Mar. Freshw. Res. 2017, 68, 1788. [Google Scholar] [CrossRef]
- Will-Wolf, S.; Geiser, L.H.; Neitlich, P.; Reis, A.H. Forest lichen communities and environment—How consistent are relationships across scales? J. Veg. Sci. 2006, 17, 171–184. [Google Scholar] [CrossRef]
- Wolseley, P.A.; Stofer, S.; Mitchell, R.; Truscott, A.-M.; Vanbergen, A.; Chimonides, J.; Scheidegger, C. Variation of lichen communities with landuse in Aberdeenshire, UK. Lichenologist 2006, 38, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, G.; St. Clair, L.L. The lichen flora of Southwestern Colorado. Evansia 2009, 26, 102–123. [Google Scholar] [CrossRef]
- Rushforth, S.R.; St. Clair, L.L.; Brotherson, J.D.; Nebeker, G.T. Lichen Community Structure in Zion National Park. Bryologist 1982, 85, 185–192. [Google Scholar] [CrossRef]
- Newberry, C.C. Lichens of the Uinta Mountains and Adjacent Intermountain North America; Brigham Young University: Provo, UT, USA, 1991. [Google Scholar]
- Rajvanshi, F.; St. Clair, L.L.; Webb, B.L.; Newberry, C.C. The terricolous lichen flora of the San Rafael Swell, Emery County, Utah, U.S.A. In Lichenographia Thomsoniana: North American Lichenology in Honor of John W. Thomson; Glenn, M., Cole, M.S., Dirig, R., Harris, R.C., Eds.; Mycotaxon LTD: Ithaca, NY, USA, 1998; pp. 399–406. [Google Scholar]
- Jackson, H.B.; Leavitt, S.D.; Krebs, T.; St. Clair, L.L. Lichen flora of the eastern Mojave Desert: Blackrock Arizona, Mojave County, Arizona, USA. Evansia 2005, 22, 30–38. [Google Scholar] [CrossRef]
- Leavitt, S.D.; St. Clair, L.L. Lichens of Boulder Mountain Plateau, Wayne, County, Utah, USA. Evansia 2008, 26, 85–89. [Google Scholar] [CrossRef]
- Robison, A.; Baugh, M.; Muggia, L.; Leavitt, S.D. Fruticose Lichen Communities at the Edge: Distribution and Diversity in a Desert Sky Island on the Colorado Plateau. Conservation 2022, 2, 550–565. [Google Scholar] [CrossRef]
- Carter, O.; Kropp, B.; Noell, N.; Hollinger, J.; Baker, G.; Tuttle, A.; St. Clair, L.L.; Leavitt, S.D. A Preliminary Checklist of the Lichens in Great Basin National Park, Nevada, USA. Evansia 2019, 36, 72–91. [Google Scholar] [CrossRef]
- Geiser, L.H.; Neitlich, P.N. Air pollution and climate gradients in western Oregon and Washington indicated by epiphytic macrolichens. Environ. Pollut. 2007, 145, 203–218. [Google Scholar] [CrossRef]
- Naidoo, K.; Steenkamp, E.T.; Coetzee, M.P.A.; Wingfield, M.J.; Wingfield, B.D. Concerted Evolution in the Ribosomal RNA Cistron. PLoS ONE 2013, 8, e59355. [Google Scholar] [CrossRef] [Green Version]
- Lindner, D.L.; Carlsen, T.; Henrik Nilsson, R.; Davey, M.; Schumacher, T.; Kauserud, H. Employing 454 amplicon pyrosequencing to reveal intragenomic divergence in the internal transcribed spacer rDNA region in fungi. Ecol. Evol. 2013, 3, 1751–1764. [Google Scholar] [CrossRef]
- Lücking, R.; Forno, M.D.; Moncada, B.; Coca, L.F.; Vargas-Mendoza, L.Y.; Aptroot, A.; Arias, L.J.; Besal, B.; Bungartz, F.; Cabrera-Amaya, D.M.; et al. Turbo-taxonomy to assemble a megadiverse lichen genus: Seventy new species of Cora (Basidiomycota: Agaricales: Hygrophoraceae), honouring David Leslie Hawksworth’s seventieth birthday. Fungal Divers. 2017, 84, 139–207. [Google Scholar] [CrossRef]
- Collins, R.A.; Cruickshank, R.H. The seven deadly sins of DNA barcoding. Mol. Ecol. Resour. 2012, 13, 969–975. [Google Scholar] [CrossRef]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to fail at species delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef]
- Tan, D.; Ang, Y.; Lim, G.; Ismail, M.; Meier, R. From ‘cryptic species’ to integrative taxonomy: An iterative process involving DNA sequences, morphology, and behaviour leads to the resurrection of Sepsis pyrrhosoma (Sepsidae: Diptera). Zool. Scr. 2009, 39, 51–61. [Google Scholar] [CrossRef]
- de Queiroz, K. Species Concepts and Species Delimitation. Syst. Biol. 2007, 56, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spribille, T. Relative symbiont input and the lichen symbiotic outcome. Curr. Opin. Plant Biol. 2018, 44, 57–63. [Google Scholar] [CrossRef]
- Printzen, C. Lichen Systematics: The Role of Morphological and Molecular Data to Reconstruct Phylogenetic Relationships. In Progress in Botany 71; Springer: Berlin/Heidelberg, Germany, 2010; pp. 233–275. [Google Scholar]
- Lumbsch, H.T.; Leavitt, S.D. Goodbye morphology? A paradigm shift in the delimitation of species in lichenized fungi. Fungal Divers. 2011, 50, 59–72. [Google Scholar] [CrossRef]
- Hendrich, L.; Morinière, J.; Haszprunar, G.; Hebert, P.D.N.; Hausmann, A.; Köhler, F.; Balke, M. A comprehensive DNA barcode database for Central European beetles with a focus on Germany: Adding more than 3500 identified species to BOLD. Mol. Ecol. Resour. 2015, 15, 795–818. [Google Scholar] [CrossRef] [PubMed]
- Radulovici, A.E.; Sainte-Marie, B.; Dufresne, F. DNA barcoding of marine crustaceans from the Estuary and Gulf of St Lawrence: A regional-scale approach. Mol. Ecol. Resour. 2009, 9, 181–187. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Name | Level 3 and 4 Ecoregions; Site Description; Latitude, Longitude, Altitude; Collection Date. | Sample Type | Species/OTUs |
|---|---|---|---|
| Brigham Young University (Utah) | Central Basin and Range/Moist Wasatch Front Footslopes Utah County, Brigham Young University Provo Campus, on cement wall and stairway, west of the McKay Building; 40.247, −111.652, 1400 m above sea level (m.a.s.l.); 5 June 2019. | Bulk | 65/94 |
| Broom Canyon (Nevada) | Central Basin and Range/Carbonate Woodland Zone Nye County, Humboldt-Toiyabe National Forest, White Pine Range (Currant Mountain Wilderness), Ely Ranger District, east of Railroad Valley, at mouth of Broom Canyon, west-facing slope of White Pine Peak, west side of Currant Mountain Wilderness Area; 38.890, −115.500, 2063 m.a.s.l.; 29 June 2011. | Bulk | 146/171 |
| Moore’s Creek (Nevada) | Central Basin and Range/Central Nevada Mid-Slope Woodland and Brushland Nye County, Humboldt-Toiyabe National Forest, Alta Toquima Wilderness, Moore’s Creek Trailhead; 38.860, −116.934, 2330 m.a.s.l.; 7 August 2013. | Vouchers | 98/125 |
| Mosquito Creek (Nevada) | Central Basin and Range/Central Nevada Mid-Slope Woodland and Brushland Nye County, Humboldt-Toiyabe National Forest, Table Mountain (Table Mountain Wilderness & vicinity), near boundary of Table Mountain Wilderness Area, along USFS Road No. 4409b, at Mosquito Creek Trailhead; 38.807, −116.682, 2210 m.a.s.l.; 8 August 2013. | Vouchers | 115/145 |
| Paul Bunyan’s Woodpile (Utah) | Central Basin and Range/Woodland- and Shrub-Covered Low Mountains Juab County, basalt dike ‘Paul Bunyan’s Woodpile’, near Jericho Junction; 39.767, −112.115, 2045 m.a.s.l.; 29 April 2019. | Bulk | 144/188 |
| Pine Creek (Utah) | Colorado Plateaus/Escarpments Garfield Co., Dixie National Forest, Box Death Hollow Wilderness Area, ~15 km north of Escalante along Hell’s Backbone Road (USFS Road No. 153), ~1.5 km north of “Box Trailhead” along USFS Trail No. 4009, along Pine Creek (collections made in riparian habitat along Pine Creek and surrounding sandstone outcrops); 37.865, −111.634, 1970 m.a.s.l.; 13 July 2015. | Vouchers | 112/135 |
| Death Hollow (Utah) | Wasatch and Uinta Mountains/High Plateaus/Wasatch and Uinta Mountains Garfield County, Dixie National Forest, Box Death Hollow Wilderness Area, at sandstone ridge south of head of Death Hollow, ~0.5 km southwest of Box Death Hollow Bridge, along Hell’s Backbone Road (USFS Road No. 153); 37.966, −111.599, 2661 m.a.s.l.; 13 July 2015. | Vouchers | 100/121 |
| Delano Peak (Utah) | Wasatch and Uinta Mountains/Alpine Zone Beaver/Piute Counties, Fish Lake National Forest, above tree line in alpine steppe habitat, vicinity of Delano Peak; 38.370, −112.376, 3650 m.a.s.l.; 16 September 2017. | Vouchers | 123/159 |
| Harmon Creek (Utah) | Wasatch and Uinta Mountains/Semiarid Foothills Washington County, Dixie National Forest, Pine Valley Mountains Wilderness Area, west of USFS Road No. 037, along USFS Trail No. 3028, vicinity of Harmon Creek Trailhead (riparian habitat along Harmon Creek); 37.364, −113.3518, 1764 m.ASL; 9 July 2015. | Vouchers | 80/103 |
| Quail Creek (Utah) | Wasatch and Uinta Mountains/Semiarid Foothills Washington County, Dixie National Forest, Cottonwood Forest Wilderness Area, Water Canyon, along Quail Creek, east of USFS Road No. 031, (Oak Grove Road), at spur road—USFS Road No. 4059, immediately south of private land (Sagewood Ranches); collections were made along Quail Creek and upslope in a Pinyon-Juniper woodland; 37.255, −113.427, 1212 m.a.s.l.; 11 July 2015. | Vouchers | 103/122 |
| Whipple Creek (Utah) | Wasatch and Uinta Mountains/High Plateaus Washington County, Dixie National Forest, Pine Valley Mountains Wilderness Area, east of Pine Valley, along Whipple Trail (USFS Trail No. 3025), east of wilderness boundary (riparian habitat along Middle Fork of Santa Clara River and upland Gambel oak-Mountain mahogany habitat); 37.368, −113.452, 2230 m.a.s.l.; 10 July 2015. | Vouchers | 96/123 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerr, M.; Leavitt, S.D. A Custom Regional DNA Barcode Reference Library for Lichen-Forming Fungi of the Intermountain West, USA, Increases Successful Specimen Identification. J. Fungi 2023, 9, 741. https://doi.org/10.3390/jof9070741
Kerr M, Leavitt SD. A Custom Regional DNA Barcode Reference Library for Lichen-Forming Fungi of the Intermountain West, USA, Increases Successful Specimen Identification. Journal of Fungi. 2023; 9(7):741. https://doi.org/10.3390/jof9070741
Chicago/Turabian StyleKerr, Michael, and Steven D. Leavitt. 2023. "A Custom Regional DNA Barcode Reference Library for Lichen-Forming Fungi of the Intermountain West, USA, Increases Successful Specimen Identification" Journal of Fungi 9, no. 7: 741. https://doi.org/10.3390/jof9070741
APA StyleKerr, M., & Leavitt, S. D. (2023). A Custom Regional DNA Barcode Reference Library for Lichen-Forming Fungi of the Intermountain West, USA, Increases Successful Specimen Identification. Journal of Fungi, 9(7), 741. https://doi.org/10.3390/jof9070741