
Development of Poly(sorbitol adipate)-g-poly(ethylene glycol) Mono Methyl Ether-Based Hydrogel Matrices for Model Drug Release

 , , ,
, , ,  and
and
Abstract
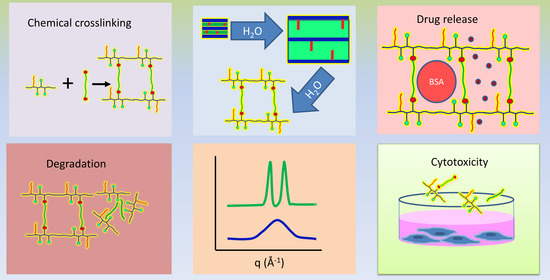
:
1. Introduction
2. Results and Discussion
2.1. Polymers and PSA-g-mPEG Hydrogel Syntheses
2.2. Stability and Degradation Study of PSA and PSA-g-mPEG
2.3. Sol-Gel Fraction of PSA-g-mPEG Hydrogels
2.4. Swelling Studies of PSA-g-mPEG Hydrogels
2.5. Temperature-Dependent Swelling Behavior of PSA-g-mPEG Hydrogels
2.6. Physical Structural Parameters of PSA-g-mPEG Hydrogels
2.7. X-ray Diffraction
2.8. Loading and Release Experiment of BSA-TMR and DY-781 from PSA-g-mPEG Hydrogels
2.9. Cytotoxicity of PSA-g-mPEG Hydrogels
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Polymers and Hydrogel Syntheses
4.3. Polymer Degradation/Stability Study
4.4. Sol-Gel Fraction of PSA-g-mPEG Hydrogels
4.5. Swelling Studies
4.6. Structural Parameters of the PSA-g-mPEG Hydrogels
4.7. X-ray Diffraction (XRD) of PSA-g-mPEG Hydrogels
4.8. Loading Study of the BSA-TMR and DY-781 into Hydrogel Matrices
4.9. Release Study of the BSA-TMR and DY-781
4.10. Cytotoxicity Study of PSA-g-mPEG Hydrogels
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doppalapudi, S.; Jain, A.; Khan, W.; Domb, A.J. Biodegradable polymers—An overview. Polym. Adv. Technol. 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Kesharwani, P.; Jain, K.; Jain, N.K. Dendrimer as nanocarrier for drug delivery. Prog. Polym. Sci. 2014, 39, 268–307. [Google Scholar] [CrossRef]
- Thakur, V.K.; Thakur, M.K.; Kessler, M.R. Handbook of Composites from Renewable Materials, Structure and Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2016; Volume 1, ISBN 1119224233. [Google Scholar]
- Surendren, A.; Cheekuramelli, N.S.; Magisetty, R.P. Biodegradable polymer blends for tissue engineering. In Biodegradable Polymers, Blends and Composites; Elsevier: Amsterdam, The Netherlands, 2022; pp. 591–609. [Google Scholar]
- Kesharwani, P.; Prajapati, S.K.; Jain, A.; Mody, N.; Sharma, S. A glimpse of biomedical application potential of biodegradable polymers for anticancer drug delivery. In Polymeric Biomaterials for Healthcare Applications; Elsevier: Amsterdam, The Netherlands, 2022; pp. 211–234. [Google Scholar]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Huang, P.-K.; Law, W.-C.; Chu, C.-H.; Chen, N.-T.; Lo, L.-W. Biodegradable polymers for gene-delivery applications. Int. J. Nanomed. 2020, 15, 2131–2150. [Google Scholar] [CrossRef]
- Bose, R.J.C.; Kim, M.; Chang, J.H.; Paulmurugan, R.; Moon, J.J.; Koh, W.-G.; Lee, S.-H.; Park, H. Biodegradable polymers for modern vaccine development. J. Ind. Eng. Chem. 2019, 77, 12–24. [Google Scholar] [CrossRef]
- Prajapati, S.K.; Jain, A.; Jain, A.; Jain, S. Biodegradable polymers and constructs: A novel approach in drug delivery. Eur. Polym. J. 2019, 120, 109191. [Google Scholar] [CrossRef]
- Kulkarni, R.K.; Moore, E.G.; Hegyeli, A.F.; Leonard, F. Biodegradable poly (lactic acid) polymers. J. Biomed. Mater. Res. 1971, 5, 169–181. [Google Scholar] [CrossRef]
- Zlomke, C.; Barth, M.; Mäder, K. Polymer degradation induced drug precipitation in PLGA implants—Why less is sometimes more. Eur. J. Pharm. Biopharm. 2019, 139, 142–152. [Google Scholar] [CrossRef]
- Mante, A.; Heider, M.; Zlomke, C.; Mäder, K. PLGA nanoparticles for peroral delivery: How important is pancreatic digestion and can we control it? Eur. J. Pharm. Biopharm. 2016, 108, 32–40. [Google Scholar] [CrossRef]
- Kempe, S.; Mäder, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef]
- Wan, J.-P.; Yang, Y.-Y.; Chung, T.-S.; Tan, D.; Ng, S.; Heller, J. POE–PEG–POE triblock copolymeric microspheres containing protein: II. Polymer erosion and protein release mechanism. J. Control. Release 2001, 75, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Pack, D.W.; Klibanov, A.M.; Langer, R. Visual evidence of acidic environment within degrading poly (lactic-co-glycolic acid)(PLGA) microspheres. Pharm. Res. 2000, 17, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Mäder, K.; Gallez, B.; Liu, K.J.; Swartz, H.M. Non-invasive in vivo characterization of release processes in biodegradable polymers by low-frequency electron paramagnetic resonance spectroscopy. Biomaterials 1996, 17, 457–461. [Google Scholar] [CrossRef]
- Wersig, T.; Hacker, M.C.; Kressler, J.; Mäder, K. Poly (glycerol adipate)–indomethacin drug conjugates–synthesis and in vitro characterization. Int. J. Pharm. 2017, 531, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Vert, M. Aliphatic polyesters: Great degradable polymers that cannot do everything. Biomacromolecules 2005, 6, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.H.; Hussain, H.; Prehm, M.; Baumeister, U.; Meister, A.; Hause, G.; Busse, K.; Mäder, K.; Kressler, J. Synthesis of poly(glycerol adipate)-g-oleate and its ternary phase diagram with glycerol monooleate and water. Eur. Polym. J. 2017, 91, 162–175. [Google Scholar] [CrossRef]
- Seyednejad, H.; Ghassemi, A.H.; Van Nostrum, C.F.; Vermonden, T.; Hennink, W.E. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J. Control. Release 2011, 152, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Kimura, S. Enzymatic Polymerization. Chem. Rev. 2001, 101, 3793–3818. [Google Scholar] [CrossRef]
- Métrai, G.; Wentland, J.; Thomann, Y.; Tiller, J.C. Biodegradable poly(ester hydrazide)s via enzymatic polymerization. Macromol. Rapid Commun. 2005, 26, 1330–1335. [Google Scholar] [CrossRef]
- Rashid, H.; Golitsyn, Y.; Bilal, M.H.; Mäder, K.; Reichert, D.; Kressler, J. Polymer networks synthesized from poly(Sorbitol adipate) and functionalized poly(ethylene glycol). Gels 2021, 7, 22. [Google Scholar] [CrossRef]
- Gross, R.A.; Ganesh, M.; Lu, W. Enzyme-catalysis breathes new life into polyester condensation polymerizations. Trends Biotechnol. 2010, 28, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Loos, K. Enzymatic synthesis of biobased polyesters and polyamides. Polymers 2016, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Douka, A.; Vouyiouka, S.; Papaspyridi, L.-M.; Papaspyrides, C.D. A review on enzymatic polymerization to produce polycondensation polymers: The case of aliphatic polyesters, polyamides and polyesteramides. Prog. Polym. Sci. 2018, 79, 1–25. [Google Scholar] [CrossRef]
- Kobayashi, S. Lipase-catalyzed polyester synthesis–a green polymer chemistry. Proc. Japan Acad. Ser. B 2010, 86, 338–365. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Enzymatic ring-opening polymerization and polycondensation for the green synthesis of polyesters. Polym. Adv. Technol. 2015, 26, 677–686. [Google Scholar] [CrossRef]
- Taresco, V.; Suksiriworapong, J.; Creasey, R.; Burley, J.C.; Mantovani, G.; Alexander, C.; Treacher, K.; Booth, J.; Garnett, M.C. Properties of acyl modified poly(glycerol-adipate) comb-like polymers and their self-assembly into nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3267–3278. [Google Scholar] [CrossRef] [PubMed]
- Wersig, T.; Krombholz, R.; Janich, C.; Meister, A.; Kressler, J.; Mäder, K. Indomethacin functionalised poly(glycerol adipate) nanospheres as promising candidates for modified drug release. Eur. J. Pharm. Sci. 2018, 123, 350–361. [Google Scholar] [CrossRef]
- Weiss, V.M.; Naolou, T.; Hause, G.; Kuntsche, J.; Kressler, J.; Mäder, K. Poly(glycerol adipate)-fatty acid esters as versatile nanocarriers: From nanocubes over ellipsoids to nanospheres. J. Control. Release 2012, 158, 156–164. [Google Scholar] [CrossRef]
- Naolou, T.; Meister, A.; Schöps, R.; Pietzsch, M.; Kressler, J. Synthesis and characterization of graft copolymers able to form polymersomes and worm-like aggregates. Soft Matter 2013, 9, 10364–10372. [Google Scholar] [CrossRef]
- Steiner, J.; Alaneed, R.; Kressler, J.; Mäder, K. Fatty acid-modified poly(glycerol adipate) microparticles for controlled drug delivery. J. Drug Deliv. Sci. Technol. 2021, 61, 102206. [Google Scholar] [CrossRef]
- Tavakol, M.; Vasheghani-Farahani, E.; Dolatabadi-Farahani, T.; Hashemi-Najafabadi, S. Sulfasalazine release from alginate-N,O-carboxymethyl chitosan gel beads coated by chitosan. Carbohydr. Polym. 2009, 77, 326–330. [Google Scholar] [CrossRef]
- Khorram, M.; Vasheghani-Farahani, E.; Dinarvand, R. Preparation of poly(-isopropylacrylamide) hollow beads as reservoir drug delivery systems. J. Control. Release 2006, 116, e31–e33. [Google Scholar] [CrossRef] [PubMed]
- Munim, S.A.; Raza, Z.A. Poly(lactic acid) based hydrogels: Formation, characteristics and biomedical applications. J. Porous Mater. 2019, 26, 881–901. [Google Scholar] [CrossRef]
- Rahmani, F.; Atabaki, R.; Behrouzi, S.; Mohamadpour, F.; Kamali, H. The recent advancement in the PLGA-based thermo-sensitive hydrogel for smart drug delivery. Int. J. Pharm. 2022, 631, 122484. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Chen, B. Modulating the Properties of Poly (glycerol sebacate)-Based Polyurethane Hydrogels Using an Organoclay. ACS Biomater. Sci. Eng. 2022, 8, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.H.; Prehm, M.; Njau, A.E.; Samiullah, M.H.; Meister, A.; Kressler, J. Enzymatic synthesis and characterization of hydrophilic sugar based polyesters and their modification with stearic acid. Polymers 2016, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Uyama, H. Enzymatic Polymerization. Future Dir. Biocatal. 2007, 101, 205–251. [Google Scholar] [CrossRef]
- Hu, J.; Gao, W.; Kulshrestha, A.; Gross, R.A. “Sweet polyesters”: Lipase-catalyzed condensation—Polymerizations of alditols. Macromolecules 2006, 39, 6789–6792. [Google Scholar] [CrossRef]
- D’souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG—A versatile conjugating ligand for drugs and drug delivery systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Mittag, J.J.; Trutschel, M.-L.; Kruschwitz, H.; Mäder, K.; Buske, J.; Garidel, P. Characterization of radicals in polysorbate 80 using electron paramagnetic resonance (EPR) spectroscopy and spin trapping. Int. J. Pharm. X 2022, 4, 100123. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-C.; Wang, H.-E.; Lin, W.-W.; Roffler, S.R.; Cheng, T.-C.; Su, Y.-C.; Li, J.-J.; Chen, C.-C.; Huang, C.-H.; Chen, B.-M. Pre-existing anti-polyethylene glycol antibody reduces the therapeutic efficacy and pharmacokinetics of PEGylated liposomes. Theranostics 2018, 8, 3164. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, S.; Madras, G. Synthesis and degradation of sorbitol-based polymers. J. Appl. Polym. Sci. 2011, 121, 2861–2869. [Google Scholar] [CrossRef]
- Swainson, S.M.E.; Taresco, V.; Pearce, A.K.; Clapp, L.H.; Ager, B.; McAllister, M.; Bosquillon, C.; Garnett, M.C. Exploring the enzymatic degradation of poly(glycerol adipate). Eur. J. Pharm. Biopharm. 2019, 142, 377–386. [Google Scholar] [CrossRef]
- Lv, A.; Cui, Y.; Du, F.-S.; Li, Z.-C. Thermally degradable polyesters with tunable degradation temperatures via postpolymerization modification and intramolecular cyclization. Macromolecules 2016, 49, 8449–8458. [Google Scholar] [CrossRef]
- Steiner, J. Fatty acid-modified poly (glycerol adipate) as a versatile matrix for parenteral depot formulations. Ph.D. Dissertation, Martin-Luther-University Halle-Wittenberg, Halle (Saale), Germany, 7 July 2022. [Google Scholar]
- Griffith, L.G. Polymeric biomaterials. Acta Mater. 2000, 48, 263–277. [Google Scholar] [CrossRef]
- Merkli, A.; Tabatabay, C.; Gurny, R.; Heller, J. Biodegradable polymers for the controlled release of ocular drugs. Prog. Polym. Sci. 1998, 23, 563–580. [Google Scholar] [CrossRef]
- Baird, J.A.; Olayo-Valles, R.; Rinaldi, C.; Taylor, L.S. Effect of molecular weight, temperature, and additives on the moisture sorption properties of polyethylene glycol. J. Pharm. Sci. 2010, 99, 154–168. [Google Scholar] [CrossRef]
- Chen, S.; Liu, M.; Jin, S.; Wang, B. Preparation of ionic-crosslinked chitosan-based gel beads and effect of reaction conditions on drug release behaviors. Int. J. Pharm. 2008, 349, 180–187. [Google Scholar] [CrossRef]
- Le Ouay, B.; Uemura, T. Polymer in MOF Nanospace: From Controlled Chain Assembly to New Functional Materials. Isr. J. Chem. 2018, 58, 995–1009. [Google Scholar] [CrossRef]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Buenger, D.; Topuz, F.; Groll, J. Hydrogels in sensing applications. Prog. Polym. Sci. 2012, 37, 1678–1719. [Google Scholar] [CrossRef]
- Lowman, A.M.; Peppas, N.A.; Morishita, M.; Nagai, T. Novel Bioadhesive Complexation Networks for Oral Protein Drug Delivery. In ACS Symposium Series; ACS Publications: Washington, DC, USA, 1998; Volume 709, pp. 156–164. ISBN 1947-5918. [Google Scholar]
- Truong, V.; Blakey, I.; Whittaker, A.K. Hydrophilic and amphiphilic polyethylene glycol-based hydrogels with tunable degradability prepared by “click” chemistry. Biomacromolecules 2012, 13, 4012–4021. [Google Scholar] [CrossRef] [PubMed]
- Atta, S.; Khaliq, S.; Islam, A.; Javeria, I.; Jamil, T.; Athar, M.M.; Shafiq, M.I.; Ghaffar, A. Injectable biopolymer based hydrogels for drug delivery applications. Int. J. Biol. Macromol. 2015, 80, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Anseth, K.S. Release behavior of high molecular weight solutes from poly(ethylene glycol)-based degradable networks. Macromolecules 2000, 33, 2509–2515. [Google Scholar] [CrossRef]
- Graham, N.B.; Zulfiqar, M. Interaction of poly(ethylene oxide) with solvents: 3. Synthesis and swelling in water of crosslinked poly(ethylene glycol) urethane networks. Polymer 1989, 30, 2130–2135. [Google Scholar] [CrossRef]
- Gnanou, Y.; Hild, G.; Bastide, J.; Rempp, P. Hydrophilic polyurethane networks exhibiting anisotropic swelling behaviour. J. Polym. Mater. 1987, 4, 123–130. [Google Scholar]
- Iza, M.; Stoianovici, G.; Viora, L.; Grossiord, J.L.; Couarraze, G. Hydrogels of poly(ethylene glycol): Mechanical characterization and release of a model drug. J. Control. Release 1998, 52, 41–51. [Google Scholar] [CrossRef]
- Hocine, S.; Li, M.H. Thermoresponsive self-assembled polymer colloids in water. Soft Matter 2013, 9, 5839–5861. [Google Scholar] [CrossRef]
- de Vringer, T.; Joosten, J.G.H.; Junginger, H.E. A study of the hydration of polyoxyethylene at low temperatures by differential scanning calorimetry. Colloid Polym. Sci. 1986, 264, 623–630. [Google Scholar] [CrossRef]
- Graham, N.B.; Chen, C.F. Interaction of poly(ethylene oxide) with solvent—5. The densities of water/poly(ethylene glycol) mixtures. Eur. Polym. J. 1993, 29, 149–151. [Google Scholar] [CrossRef]
- Antonsen, K.P.; Hoffman, A.S. Water Structure of PEG Solutions by Differential Scanning Calorimetry Measurements. In Poly(ethylene glycol) Chemistry; Springer: Boston, MA, USA, 1992; pp. 15–28. [Google Scholar] [CrossRef]
- Lin, C.C.; Metters, A.T. Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 2006, 58, 1379–1408. [Google Scholar] [CrossRef] [PubMed]
- Munch, J.P.; Candau, S.; Herz, J.; Hild, G. Inelastic Light Scattering By Gel Modes in Semi-Dilute Polymer Solutions and Permanent Networks at Equilibrium Swollen State. J. Phys. 1977, 38, 971–976. [Google Scholar] [CrossRef]
- Canal, T.; Peppas, N.A. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res. 1989, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Munoz-Pinto, D.; Qu, X.; Hou, Y.; Grunlan, M.A.; Hahn, M.S. Influence of hydrogel mechanical properties and mesh size on vocal fold fibroblast extracellular matrix production and phenotype. Acta Biomater. 2008, 4, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- am Ende, M.T.; Mikos, A.G. Diffusion-controlled delivery of proteins from hydrogels and other hydrophilic systems. In Protein Delivery: Physical Sytsem; Springer: Boston, MA, USA, 2002; pp. 139–165. [Google Scholar] [CrossRef]
- Brannon-Peppas, L.; Peppas, N.A. Equilibrium swelling behavior of pH-sensitive hydrogels. Chem. Eng. Sci. 1991, 46, 715–722. [Google Scholar] [CrossRef]
- Martens, P.; Anseth, K.S. Characterization of hydrogels formed from acrylate modified poly (vinyl alcohol) macromers. Polymer 2000, 41, 7715–7722. [Google Scholar] [CrossRef]
- Waters, D.J.; Engberg, K.; Parke-Houben, R.; Hartmann, L.; Ta, C.N.; Toney, M.F.; Frank, C.W. Morphology of photopolymerized end-linked poly(ethylene glycol) hydrogels by small-angle X-ray scattering. Macromolecules 2010, 43, 6861–6870. [Google Scholar] [CrossRef]
- Borges, F.T.P.; Papavasiliou, G.; Teymour, F. Characterizing the Molecular Architecture of Hydrogels and Crosslinked Polymer Networks beyond Flory-Rehner-I. Theory. Biomacromolecules 2020, 21, 5104–5118. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, K. Poly (oxyethylene)−water interactions: A molecular dynamics study. J. Am. Chem. Soc. 1996, 118, 8459–8469. [Google Scholar] [CrossRef]
- Takahashi, Y.; Sumita, I.; Tadokoro, H. Structural studies of polyethers. IX. Planar zigzag modification of poly(ethylene oxide). J. Polym. Sci. Part A-2 Polym. Phys. 1973, 11, 2113–2122. [Google Scholar] [CrossRef]
- Andrade, J. Hydrogels in Medicine and Pharmacy; CRC Press: Boca Raton, FL, USA, 1989; Volume 10. [Google Scholar]
- Kim, S.W.; Bae, Y.H.; Okano, T. Hydrogels: Swelling, Drug Loading, and Release. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1992, 9, 283–290. [Google Scholar]
- Gutowska, A.; Bae, Y.H.; Feijen, J.; Kim, S.W. Heparin release from thermosensitive hydrogels. J. Control. Release 1992, 22, 95–104. [Google Scholar] [CrossRef]
- Ferreira, L.; Vidal, M.M.; Gil, M.H. Evaluation of poly(2-hydroxyethyl methacrylate) gels as drug delivery systems at different pH values. Int. J. Pharm. 2000, 194, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Alaneed, R.; Golitsyn, Y.; Hauenschild, T.; Pietzsch, M.; Reichert, D.; Kressler, J. Network formation by aza-Michael addition of primary amines to vinyl end groups of enzymatically synthesized poly(glycerol adipate). Polym. Int. 2020, 70, 135–144. [Google Scholar] [CrossRef]
- Anderson, J.M. Future challenges in the in vitro and in vivo evaluation of biomaterial biocompatibility. Regen. Biomater. 2016, 3, 73–77. [Google Scholar] [CrossRef]
- Wiegand, C.; Hipler, U.-C. Evaluation of biocompatibility and cytotoxicity using keratinocyte and fibroblast cultures. Skin Pharmacol. Physiol. 2009, 22, 74–82. [Google Scholar] [CrossRef]
- Mei, Y.; Kumar, A.; Gao, W.; Gross, R.; Kennedy, S.B.; Washburn, N.R.; Amis, E.J.; Elliott, J.T. Biocompatibility of sorbitol-containing polyesters. Part I: Synthesis, surface analysis and cell response in vitro. Biomaterials 2004, 25, 4195–4201. [Google Scholar] [CrossRef]
- Turner, J.L.; Bierman, E.L. Effects of glucose and sorbitol on proliferation of cultured human skin fibroblasts and arterial smooth-muscle cells. Diabetes 1978, 27, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.H.; Alaneed, R.; Steiner, J.; Mäder, K.; Pietzsch, M.; Kressler, J. Multiple grafting to enzymatically synthesized polyesters. Methods Enzymol. 2019, 627, 57–97. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhong, W. Synthesis of propargyl-terminated heterobifunctional poly(ethylene glycol). Polymers 2010, 2, 407–417. [Google Scholar] [CrossRef]
- Ullah, K.; Ali Khan, S.; Murtaza, G.; Sohail, M.; Azizullah; Manan, A.; Afzal, A. Gelatin-based hydrogels as potential biomaterials for colonic delivery of oxaliplatin. Int. J. Pharm. 2019, 556, 236–245. [Google Scholar] [CrossRef]
- Ullah, K.; Sohail, M.; Murtaza, G.; Khan, S.A. Natural and synthetic materials based CMCh/PVA hydrogels for oxaliplatin delivery: Fabrication, characterization, In-Vitro and In-Vivo safety profiling. Int. J. Biol. Macromol. 2019, 122, 538–548. [Google Scholar] [CrossRef]
- Peppas, N.A.; Huang, Y.; Torres-Lugo, M.; Ward, J.H.; Zhang, J. Physicochemical foundations and structural design of hydrogels in medicine and biology. Annu. Rev. Biomed. Eng. 2000, 2, 9–29. [Google Scholar] [CrossRef]
- Nakajima, A.; Tanaami, K. Dissolution and Chain Dimensions of Nylon 6 in Metal Halide—Alcohol Systems. Polym. J. 1974, 5, 248–254. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogels | Q | ʋ2,s | (g·mol−1) | ξ(Å) |
|---|---|---|---|---|
| Hydrogels crosslinked with PEG-400 | 3.81 | 0.174 | 1941 | 15 |
| Hydrogels crosslinked with PEG-1000 | 6.74 | 0.109 | 3869 | 26 |
| Hydrogels crosslinked with PEG-2000 | 11.80 | 0.068 | 5772 | 38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashid, H.; Lucas, H.; Busse, K.; Kressler, J.; Mäder, K.; Trutschel, M.-L. Development of Poly(sorbitol adipate)-g-poly(ethylene glycol) Mono Methyl Ether-Based Hydrogel Matrices for Model Drug Release. Gels 2024, 10, 17. https://doi.org/10.3390/gels10010017
Rashid H, Lucas H, Busse K, Kressler J, Mäder K, Trutschel M-L. Development of Poly(sorbitol adipate)-g-poly(ethylene glycol) Mono Methyl Ether-Based Hydrogel Matrices for Model Drug Release. Gels. 2024; 10(1):17. https://doi.org/10.3390/gels10010017
Chicago/Turabian StyleRashid, Haroon, Henrike Lucas, Karsten Busse, Jörg Kressler, Karsten Mäder, and Marie-Luise Trutschel. 2024. "Development of Poly(sorbitol adipate)-g-poly(ethylene glycol) Mono Methyl Ether-Based Hydrogel Matrices for Model Drug Release" Gels 10, no. 1: 17. https://doi.org/10.3390/gels10010017
APA StyleRashid, H., Lucas, H., Busse, K., Kressler, J., Mäder, K., & Trutschel, M. -L. (2024). Development of Poly(sorbitol adipate)-g-poly(ethylene glycol) Mono Methyl Ether-Based Hydrogel Matrices for Model Drug Release. Gels, 10(1), 17. https://doi.org/10.3390/gels10010017