
Mechanically Tunable Hydrogels with Self-Healing and Shape Memory Capabilities from Thermo-Responsive Amino Acid-Derived Vinyl Polymers


Abstract
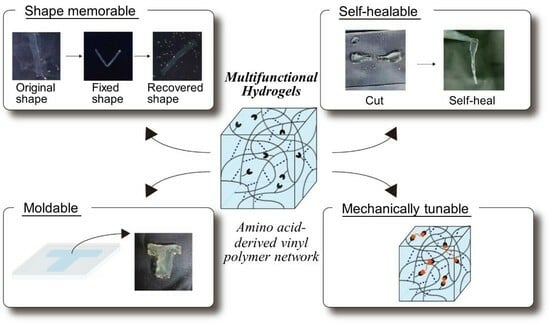
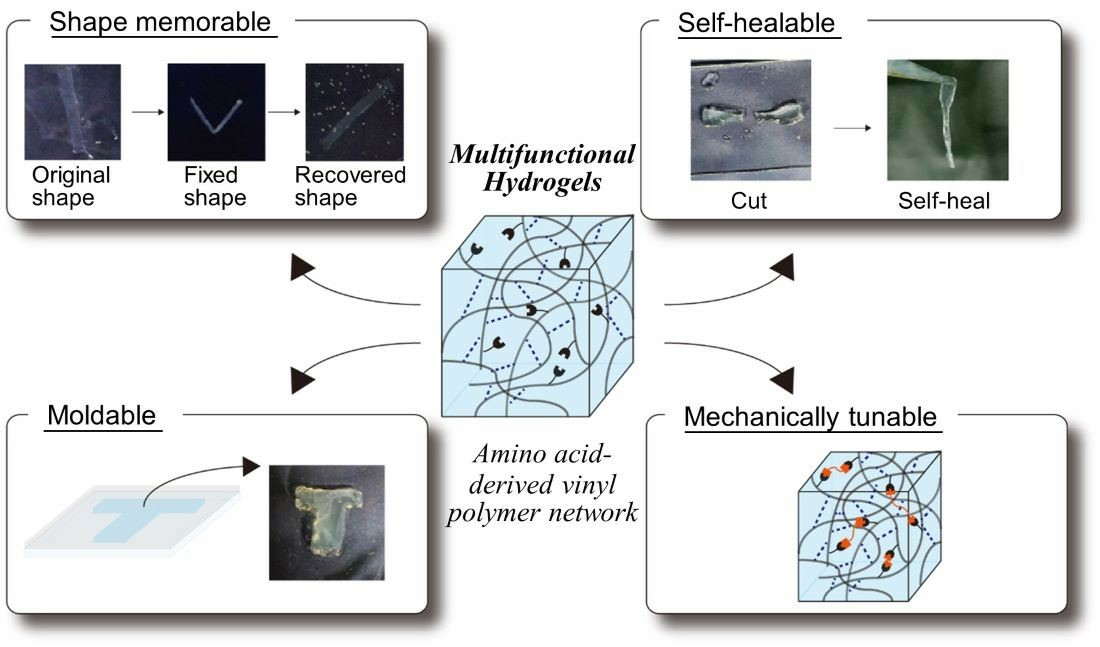
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
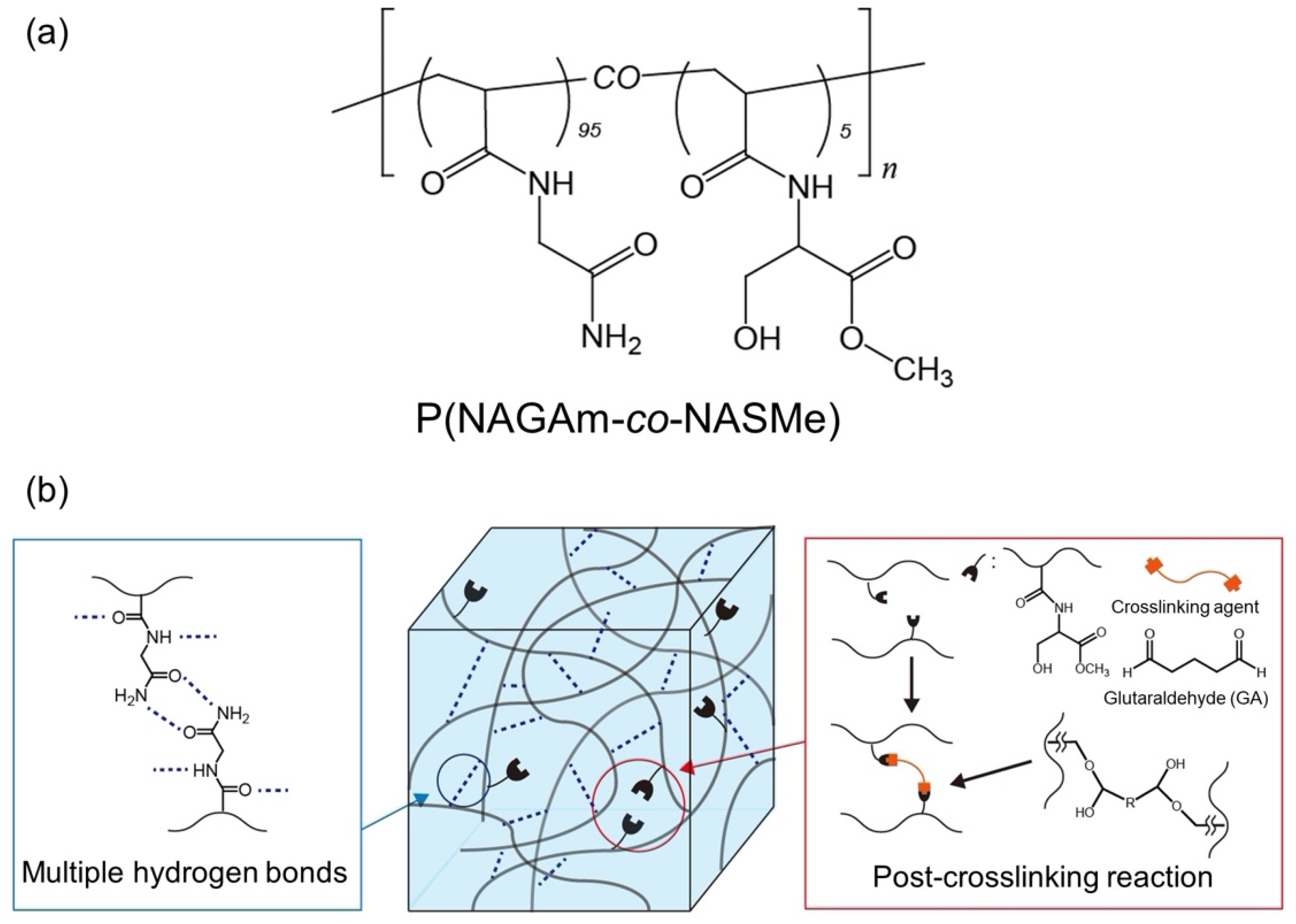
2.1. Preparation of Hydrogels from Amino Acid-Derived Vinyl Polymer
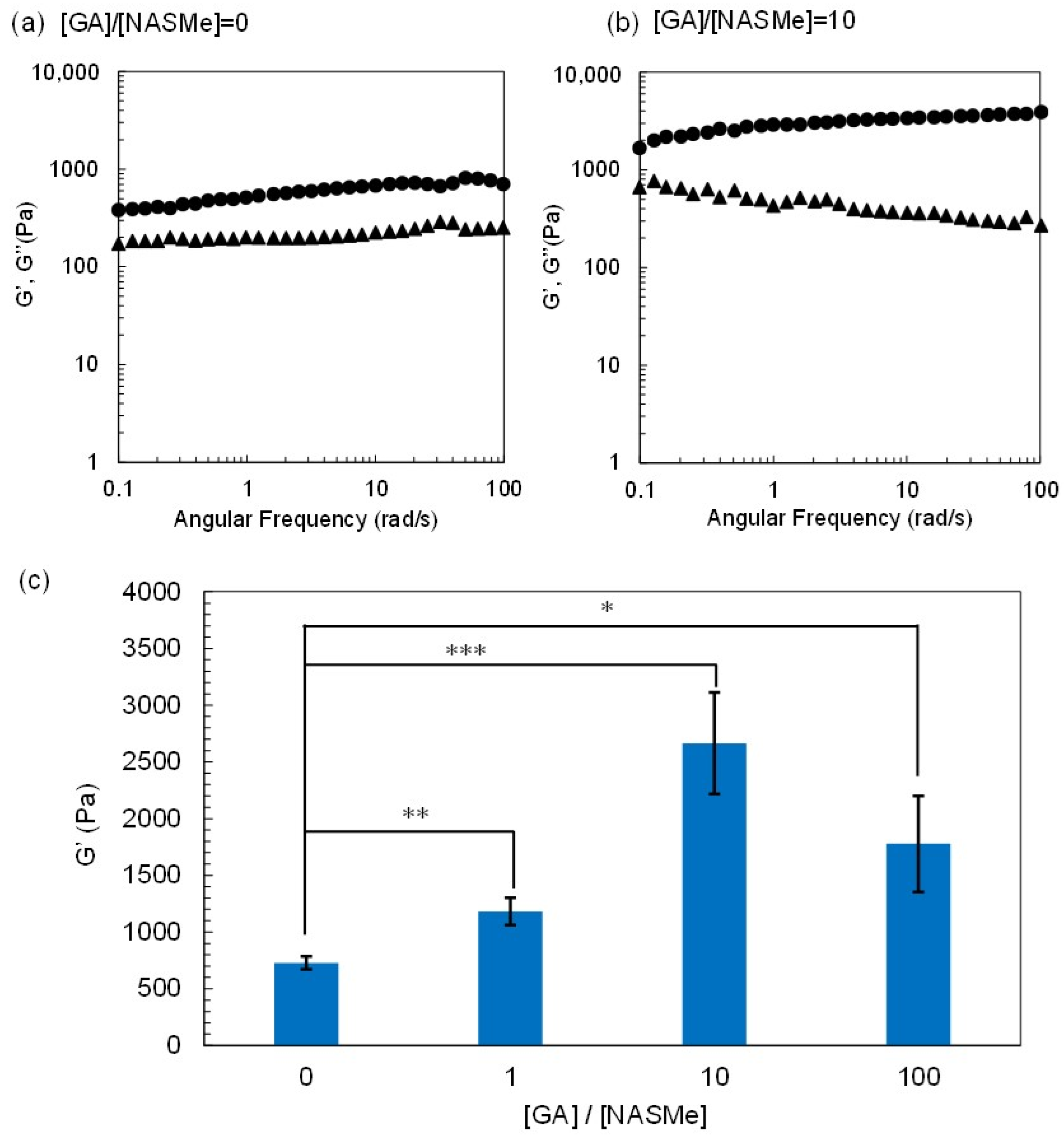
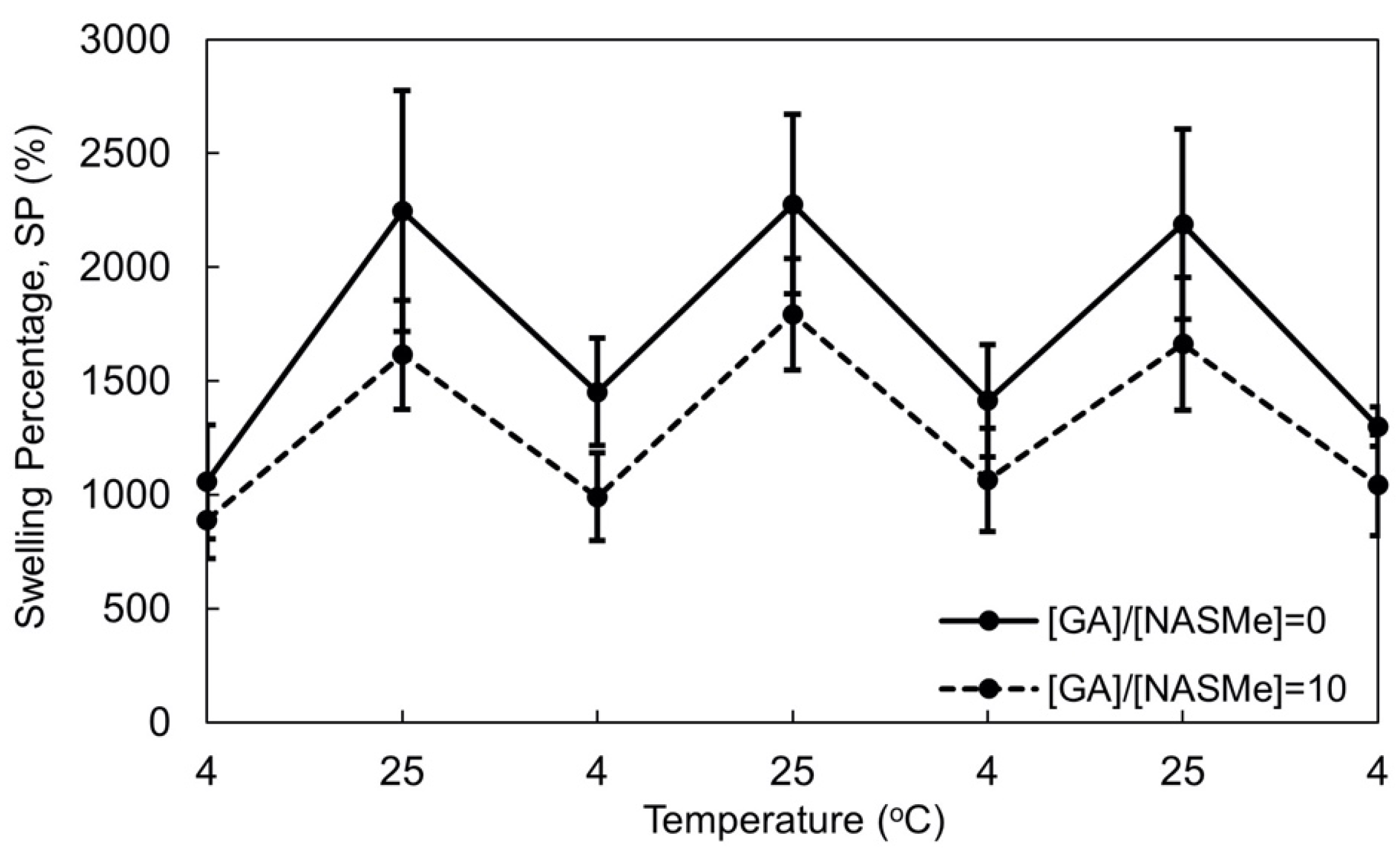
2.2. Hydrogel Property of P(NAGAm-co-NASMe) Post-Crosslinked with GA
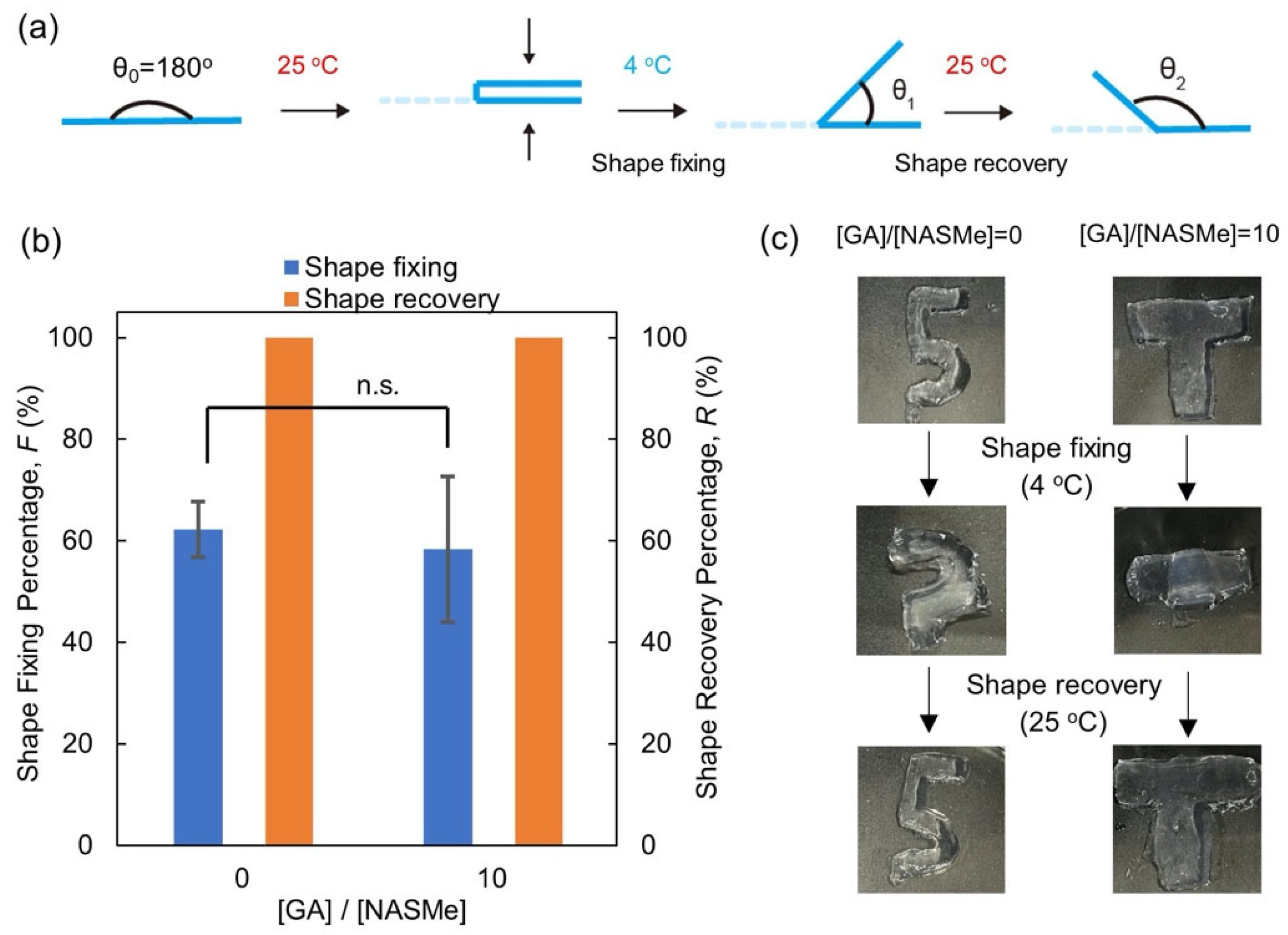
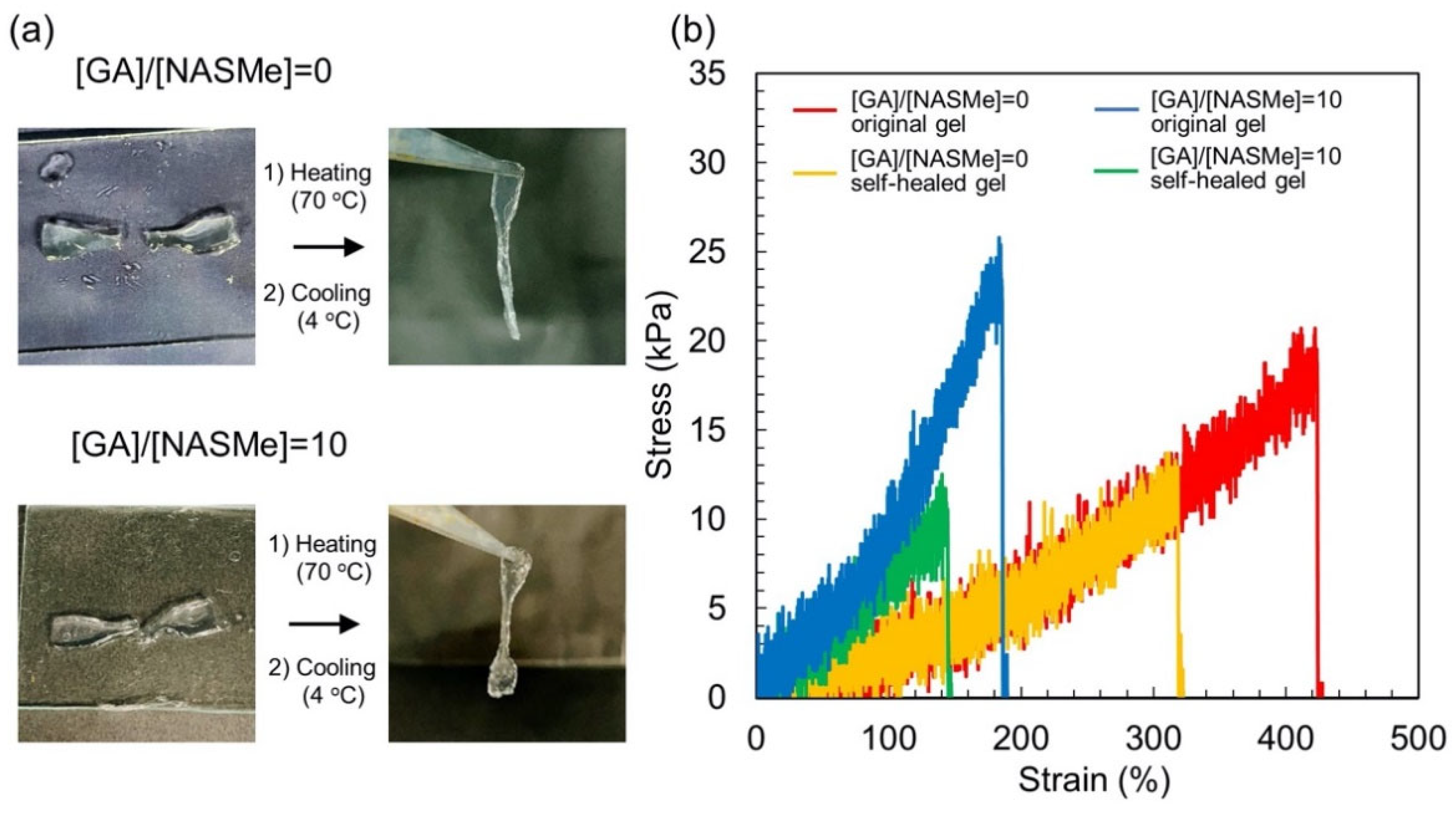
2.3. Shape-Memory and Self-Healing Behaviors of P(NAGAm-co-NASMe) Hydrogels
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Measurements
4.3. Synthesis of N-Acryloyl Glycinamide (NAGAm) and N-Acryloyl Serine Methyl Ester (NASMe)
4.4. Synthesis of P(NAGAm-co-NASMe)
4.5. Preparation of Hydrogel and Its Post-Crosslinking Reaction
4.6. Shape Memory Experiments
4.7. Swelling Behavior of Hydrogels
4.8. Self-Healing Behavior of Hydrogels
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Seliktar, D. Designing Cell-Compatible Hydrogels for Biomedical Applications. Science 2012, 336, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. Stimuli-responsive Polymers: Biomedical Applications and Challenges for Clinical Translation. Adv. Drug Deliv. Rev. 2013, 65, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, X.; Zeng, L.; Wu, L.; Guo, H.; Hourdet, D. Stimuli-responsive Toughening of Hydrogels. Chem. Mater. 2021, 33, 7633–7656. [Google Scholar] [CrossRef]
- Okumura, Y.; Ito, K. The Polyrotaxane Gel: A Topological Gel by Figure-of-Eight Cross-links. Adv. Mater. 2001, 13, 485–487. [Google Scholar] [CrossRef]
- Haque, M.A.; Kurokawa, T.; Gong, J.P. Super Tough Double Network Hydrogels and Their Application as Biomaterials. Polymer 2012, 53, 1805–1822. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly Stretchable and Tough Hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Nakahata, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Redox-responsive Self-Healing Materials Formed from Host–guest Polymers. Nat. Commun. 2011, 2, 511. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wollenberg, A.L.; O’Shea, T.M.; Cui, Y.; Zhou, Z.H.; Sofroniew, M.V.; Deming, T.J. Conformation-Directed Formation of Self-Healing Diblock Copolypeptide Hydrogels via Polyion Complexation. J. Am. Chem. Soc. 2017, 139, 15114–15121. [Google Scholar] [CrossRef]
- Talebian, S.; Mehrali, M.; Taebnia, N.; Pennisi, C.P.; Kadumudi, F.B.; Foroughi, J.; Hasany, M.; Nikkhah, M.; Akbari, M.; Orive, G.; et al. Self-Healing Hydrogels: The Next Paradigm Shift in Tissue Engineering? Adv. Sci. 2019, 6, 1801664. [Google Scholar]
- Rammal, H.; GhavamiNejad, A.; Erdem, A.; Mbeleck, R.; Nematollahi, M.; Emir Diltemiz, S.; Alem, H.; Darabi, M.A.; Ertas, Y.N.; Caterson, E.J.; et al. Advances in Biomedical Applications of Self-healing Hydrogels. Mater. Chem. Front. 2021, 5, 4368–4400. [Google Scholar] [CrossRef]
- Liang, K.; Bae, K.H.; Kurisawa, M. Recent Advances in the Design of Injectable Hydrogels for Stem Cell-based Therapy. J. Mater. Chem. B 2019, 7, 3775–3791. [Google Scholar] [CrossRef]
- Chao, Y.; Chen, Q.; Liu, Z. Smart Injectable Hydrogels for Cancer Immunotherapy. Adv. Funct. Mater. 2020, 30, 1902785. [Google Scholar] [CrossRef]
- Koga, T.; Matsuoka, T.; Morita, Y.; Higashi, N. Injectable Hydrogels Self-assembled from Oligopeptide-poly(2-methacryloyloxyethyl phosphorylcholine) Hybrid Graft Copolymers for Cell Scaffolds and Controlled Release Applications. Mater. Adv. 2021, 2, 4068–4074. [Google Scholar] [CrossRef]
- Löwenberg, C.; Balk, M.; Wischke, C.; Behl, M.; Lendlein, A. Shape-Memory Hydrogels: Evolution of Structural Principles to Enable Shape Switching of Hydrophilic Polymer Networks. Acc. Chem. Res. 2017, 50, 723–732. [Google Scholar] [CrossRef]
- Chan, B.Q.Y.; Low, Z.W.K.; Heng, S.J.W.; Chan, S.Y.; Owh, C.; Loh, X.J. Recent Advances in Shape Memory Soft Materials for Biomedical Applications. ACS Appl. Mater. Interfaces 2016, 8, 10070–10087. [Google Scholar] [CrossRef]
- Shang, J.; Le, X.; Zhang, J.; Chen, T.; Theato, P. Trends in Polymeric Shape Memory Hydrogels and Hydrogel Actuators. Polym. Chem. 2019, 10, 1036–1055. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, S.; Zhu, H.; Hu, H.; Yuan, N.; Ding, J. Self-Healing Hydrogel Sensors with Multiple Shape Memory Properties for Human Motion Monitoring. New J. Chem. 2021, 45, 314–320. [Google Scholar] [CrossRef]
- Bertsch, P.; Diba, M.; Mooney, D.J.; Leeuwenburgh, S.C.G. Self-Healing Injectable Hydrogels for Tissue Regeneration. Chem. Rev. 2023, 123, 834–873. [Google Scholar] [CrossRef]
- Guo, W.; Lu, C.-H.; Orbach, R.; Wang, F.; Qi, X.-J.; Cecconello, A.; Seliktar, D.; Willner, I. PH-Stimulated DNA Hydrogels Exhibiting Shape-Memory Properties. Adv. Mater. 2015, 27, 73–78. [Google Scholar] [CrossRef]
- Hu, X.; Vatankhah-Varnoosfaderani, M.; Zhou, J.; Li, Q.; Sheiko, S.S. Weak Hydrogen Bonding Enables Hard, Strong, Tough, and Elastic Hydrogels. Adv. Mater. 2015, 27, 6899–6905. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Liu, W. Dipole–Dipole and H-Bonding Interactions Significantly Enhance the Multifaceted Mechanical Properties of Thermoresponsive Shape Memory Hydrogels. Adv. Funct. Mater. 2015, 25, 471–480. [Google Scholar] [CrossRef]
- Feng, Z.; Zuo, H.; Gao, W.; Ning, N.; Tian, M.; Zhang, L. A Robust, Self-Healable, and Shape Memory Supramolecular Hydrogel by Multiple Hydrogen Bonding Interactions. Macromol. Rapid Commun. 2018, 39, 1800138. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Tomimori, K.; Higashi, N. Transparent, High-Strength, and Shape Memory Hydrogels from Thermo-Responsive Amino Acid-Derived Vinyl Polymer Networks. Macromol. Rapid Commun. 2020, 41, 1900650. [Google Scholar] [CrossRef] [PubMed]
- Osada, Y.; Matsuda, A. Shape Memory in Hydrogels. Nature 1995, 376, 219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Desai, M.S.; Wang, T.; Lee, S.-W. Elastin-Based Thermoresponsive Shape-Memory Hydrogels. Biomacromolecules 2020, 21, 1149–1156. [Google Scholar] [CrossRef]
- Yasin, A.; Li, H.; Lu, Z.; ur Rehman, S.; Siddiq, M.; Yang, H. A Shape Memory Hydrogel Induced by the Interactions between Metal Ions and Phosphate. Soft Matter 2014, 10, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zhang, Y.; Li, Y.; Xu, B.; Liu, W. Hydrogen Bonded and Ionically Crosslinked High Strength Hydrogels Exhibiting Ca2+-Triggered Shape Memory Properties and Volume Shrinkage for Cell Detachment. J. Mater. Chem. B 2015, 3, 6347–6354. [Google Scholar] [CrossRef]
- Miyamae, K.; Nakahata, M.; Takashima, Y.; Harada, A. Self-Healing, Expansion–Contraction, and Shape-Memory Properties of a Preorganized Supramolecular Hydrogel through Host–Guest Interactions. Angew. Chem. Int. Ed. 2015, 54, 8984–8987. [Google Scholar] [CrossRef]
- Yasin, A.; Zhou, W.; Yang, H.; Li, H.; Chen, Y.; Zhang, X. Shape Memory Hydrogel Based on a Hydrophobically-Modified Polyacrylamide (HMPAM)/α-CD Mixture via a Host-Guest Approach. Macromol. Rapid Commun. 2015, 36, 845–851. [Google Scholar] [CrossRef]
- Seuring, J.; Agarwal, S. Non-Ionic Homo- and Copolymers with H-Donor and H-Acceptor Units with an UCST in Water. Macromol. Chem. Phys. 2010, 211, 2109–2117. [Google Scholar] [CrossRef]
- Boustta, M.; Colombo, P.-E.; Lenglet, S.; Poujol, S.; Vert, M. Versatile UCST-based Thermoresponsive Hydrogels for Loco-regional Sustained Drug Delivery. J. Control. Release 2014, 174, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, W. Poly(N-Acryloyl Glycinamide): A Fascinating Polymer That Exhibits a Range of Properties from UCST to High-Strength Hydrogels. Chem. Commun. 2018, 54, 10540–10553. [Google Scholar] [CrossRef] [PubMed]
- Trappmann, B.; Gautrot, J.E.; Connelly, J.T.; Strange, D.G.T.; Li, Y.; Oyen, M.L.; Cohen Stuart, M.A.; Boehm, H.; Vogel, V.; Li, B.; et al. Extracellular-matrix Tethering Regulates Stem-Cell Fate. Nat. Mater. 2012, 11, 642–649. [Google Scholar]
- Thiele, J.; Ma, Y.; Bruekers, S.M.C.; Ma, S.; Huck, W.T.S. Designer Hydrogels for Cell Cultures: A Materials Selection Guide. Adv. Mater. 2014, 26, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Elsawy, M.A.; Wychowaniec, J.K.; Castillo Díaz, L.A.; Smith, A.M.; Miller, A.F.; Saiani, A. Controlling Doxorubicin Release from a Peptide Hydrogel through Fine-Tuning of Drug–Peptide Fiber Interactions. Biomacromolecules 2022, 23, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Kotsuchibashi, Y.; Ebara, M.; Aoyagi, T.; Narain, R. Recent Advances in Dual Temperature Responsive Block Copolymers and Their Potential as Biomedical Applications. Polymers 2016, 8, 380. [Google Scholar] [CrossRef]
- Mori, H.; Iwaya, H.; Nagai, A.; Endo, T. Controlled Synthesis of Thermoresponsive Polymers Derived from l-Proline via RAFT Polymerization. Chem. Commun. 2005, 38, 4872–4874. [Google Scholar] [CrossRef]
- Zhu, Y.; Lowe, A.B.; Roth, P.J. Postpolymerization Synthesis of (Bis)amide (co)polymers: Thermoresponsive Behavior and Self-association. Polymer 2014, 55, 4425–4431. [Google Scholar] [CrossRef]
- Higashi, N.; Sonoda, R.; Koga, T. Thermo-responsive Amino acid-based Vinyl Polymers Showing Widely Tunable LCST/UCST Behavior in Water. RSC Adv. 2015, 5, 67652–67657. [Google Scholar] [CrossRef]
- Higashi, N.; Hirata, A.; Nishimura, S.; Koga, T. Thermo-responsive Polymer Brushes on Glass Plate Prepared from a New Class of Amino Acid-derived Vinyl Monomers and Their Applications in Cell-sheet Engineering. Colloids Surf. B Biointerfaces 2017, 159, 39–46. [Google Scholar] [CrossRef]
- Bauri, K.; Nandi, M.; De, P. Amino acid-derived Stimuli-responsive Polymers and Their Applications. Polym. Chem. 2018, 9, 1257–1287. [Google Scholar] [CrossRef]
- Yamano, T.; Higashi, N.; Koga, T. Precisely Synthesized Sequence-Controlled Amino Acid–Derived Vinyl Polymers: New Insights into Thermo-Responsive Polymer Design. Macromol. Rapid Commun. 2020, 41, 1900550. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Higashi, N.; Koga, T. Unique Self-Assembly of Sequence-Controlled Amino Acid Derived Vinyl Polymer with Gradient Thermoresponsiveness along a Chain. Langmuir 2020, 36, 6550–6556. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, A.; Nishimura, S.; Higashi, N.; Koga, T. Synthesis of Amino acid-derived Vinyl Polymers with Precisely Controlled Hydropathy and Their Thermoresponsive Behavior in Water. Polym. Chem. 2023, 14, 2857–2864. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Montclare, J.K. Free-Standing Photocrosslinked Protein Polymer Hydrogels for Sustained Drug Release. Biomacromolecules 2021, 22, 1509–1522. [Google Scholar] [CrossRef]
- Tamura, A.; Lee, D.H.; Arisaka, Y.; Kang, T.W.; Yui, N. Post-Cross-Linking of Collagen Hydrogels by Caroxymethylated Polyrotaxanes for Simultaneously Improving Mechanical Strength and Cell Proliferation. ACS Biomater. Sci. Eng. 2022, 8, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhao, Y.; Xie, Q.; Fan, C.; Hilborn, J.; Dai, J. Moldable Hyaluronan Hydrogel Enabled by Dynamic Metal-Bisphosphonate Coordination Chemistry for Wound Healing. Adv. Healthc. Mater. 2018, 7, 1700973. [Google Scholar] [CrossRef] [PubMed]
- Kirchmajer, D.M.; Gorkin, R.; in het Panhuis, M. An Overview of the Suitability of Hydrogel-forming Polymers for Extrusion-based 3D-printing. J. Mater. Chem. B 2015, 3, 4105–4117. [Google Scholar]
- Kikuchi, M.; Matsumoto, H.N.; Yamada, T.; Koyama, Y.; Takakuda, K.; Tanaka, J. Glutaraldehyde Cross-linked Hydroxyapatite/Collagen Self-organized Nanocomposites. Biomaterials 2004, 25, 63–69. [Google Scholar] [CrossRef]
- Tian, Y.; Lai, J.N.; Li, C.; Sun, J.; Liu, K.; Zhao, C.; Zhang, M. Poly(N-acryloyl glycinamide-co-N-acryloxysuccinimide) Nanoparticles: Tunable Thermo-Responsiveness and Improved Bio-Interfacial Adhesion for Cell Function Regulation. ACS Appl. Mater. Interfaces 2023, 15, 7867–7877. [Google Scholar] [CrossRef] [PubMed]
- Majstorvoić, N.; Zahedtalaban, M.; Agarwal, S. Printable Poly(N-acryloyl glycinamide) Nanocomposite Hydrogel Formulations. Polym. J. 2023, 55, 1085–1095. [Google Scholar] [CrossRef]
- Hass, H.C.; Chiklis, C.K.; Moreau, R.D. Synthetic Thermally Reversible Gel Systems. III. J. Polym. Sci. A1 1970, 8, 1131–1145. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishimura, S.-n.; Sato, D.; Koga, T. Mechanically Tunable Hydrogels with Self-Healing and Shape Memory Capabilities from Thermo-Responsive Amino Acid-Derived Vinyl Polymers. Gels 2023, 9, 829. https://doi.org/10.3390/gels9100829
Nishimura S-n, Sato D, Koga T. Mechanically Tunable Hydrogels with Self-Healing and Shape Memory Capabilities from Thermo-Responsive Amino Acid-Derived Vinyl Polymers. Gels. 2023; 9(10):829. https://doi.org/10.3390/gels9100829
Chicago/Turabian StyleNishimura, Shin-nosuke, Dan Sato, and Tomoyuki Koga. 2023. "Mechanically Tunable Hydrogels with Self-Healing and Shape Memory Capabilities from Thermo-Responsive Amino Acid-Derived Vinyl Polymers" Gels 9, no. 10: 829. https://doi.org/10.3390/gels9100829
APA StyleNishimura, S. -n., Sato, D., & Koga, T. (2023). Mechanically Tunable Hydrogels with Self-Healing and Shape Memory Capabilities from Thermo-Responsive Amino Acid-Derived Vinyl Polymers. Gels, 9(10), 829. https://doi.org/10.3390/gels9100829