

Polymeric Networks Containing Amine Derivatives as Organocatalysts for Knoevenagel Reaction within Continuously Driven Microfluidic Reactors


Abstract
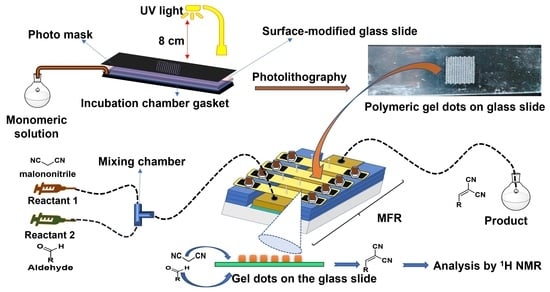
:
1. Introduction
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Analysis
4.3. Synthesis of Catalytic Monomers
4.3.1. Synthesis of tert-butyl N-(2-aminoethyl)carbamate (1)
4.3.2. Synthesis of tert-butyl N-[2-(prop-2-enoylamino)ethyl]carbamate (2)
4.3.3. Synthesis of 2-(methacryloyloxy)ethyl-4-((2-((tert-butoxycarbonyl)amino) ethyl)amino)-4-oxobutanoate (3)
4.3.4. Synthesis of N-(2-aminoethyl)prop-2-enamide. hydrochloride (4)
4.3.5. Synthesis of 2-(methacryloyloxy)ethyl-4-((2-amino) ethyl)amino)-4-oxobutanoate. hydrochloride (5)
4.4. Modification of Glass Slides
4.5. Preparation of Gel Dots Using Various Compositions of Gel Dots
4.6. Swelling Studies of Gels
4.7. Microfluidic Reactor Assembly
4.8. Microfluidic Flow Reaction
4.9. Determination of Conversion
4.10. Knoevenagel Reaction between Different Aldehydes with Malononitrile in the Continuous Flow
4.10.1. Synthesis of 2-(4-methoxybenzylidene) malononitrile (entry 1–6)
4.10.2. Synthesis of 2-benzylidenemalononitrile (entry 7–12)
4.10.3. Synthesis of 2-(2-methylpropylidene) malononitrile (entry 13–18)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Govender, T.; Arvidsson, P.I.; Maguire, G.E.M.; Kruger, H.G.; Naicker, T. Enantioselective Organocatalyzed Transformations of β-Ketoesters. Chem. Rev. 2016, 116, 9375–9437. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Notz, W.; Tanaka, F.; Barbas, C.F. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F. Amines as Catalysts: Dynamic Features and Kinetic Control of Catalytic Asymmetric Chemical Transformations to Form C-C Bonds and Complex Molecules. Chem. Rec. 2022, e202200207. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Michrowska, A.; Sulzer-Mosse, S.; List, B. The catalytic asymmetric Knoevenagel condensation. Angew. Chem. Int. Ed. Engl. 2011, 50, 1707–1710. [Google Scholar] [CrossRef]
- Dalessandro, E.V.; Collin, H.P.; Guimarães, L.G.L.; Valle, M.S.; Pliego, J.R. Mechanism of the Piperidine-Catalyzed Knoevenagel Condensation Reaction in Methanol: The Role of Iminium and Enolate Ions. J. Phys. Chem. B 2017, 121, 5300–5307. [Google Scholar] [CrossRef]
- Fadhel, A.Z.; Pollet, P.; Liotta, C.L.; Eckert, C.A. Combining the benefits of homogeneous and heterogeneous catalysis with tunable solvents and nearcritical water. Molecules 2010, 15, 8400–8424. [Google Scholar] [CrossRef] [Green Version]
- Berg, P.; Obst, F.; Simon, D.; Richter, A.; Appelhans, D.; Kuckling, D. Novel Application of Polymer Networks Carrying Tertiary Amines as a Catalyst Inside Microflow Reactors Used for Knoevenagel Reactions. Eur. J. Org. Chem. 2020, 2020, 5765–5774. [Google Scholar] [CrossRef]
- Zacuto, M.J. Synthesis of Acrylamides via the Doebner-Knoevenagel Condensation. J. Org. Chem. 2019, 84, 6465–6474. [Google Scholar] [CrossRef]
- Tokeshi, M. Applications of Microfluidic Systems in Biology and Medicine; Springer: Singapore, 2019; ISBN 978-981-13-6228-6. [Google Scholar]
- Rodrigues, T.; Schneider, P.; Schneider, G. Accessing new chemical entities through microfluidic systems. Angew. Chem. Int. Ed Engl. 2014, 53, 5750–5758. [Google Scholar] [CrossRef]
- Jas, G.; Kirschning, A. Continuous flow techniques in organic synthesis. Chemistry 2003, 9, 5708–5723. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.; Nie, Y.; Zhang, J.X.J. Microfluidics for silica biomaterials synthesis: Opportunities and challenges. Biomater. Sci. 2019, 7, 2218–2240. [Google Scholar] [CrossRef] [PubMed]
- Alias, A.B.; Mishra, S.; Pendharkar, G.; Chen, C.-S.; Liu, C.-H.; Liu, Y.-J.; Yao, D.-J. Microfluidic Microalgae System: A Review. Molecules 2022, 27, 1910. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Steinbock, O. Materials Synthesis and Catalysis in Microfluidic Devices: Prebiotic Chemistry in Mineral Membranes. ChemCatChem 2020, 12, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Carta, M.; Croad, M.; Bugler, K.; Msayib, K.J.; McKeown, N.B. Heterogeneous organocatalysts composed of microporous polymer networks assembled by Tröger’s base formation. Polym. Chem. 2014, 5, 5262. [Google Scholar] [CrossRef] [Green Version]
- Corain, B.; Zecca, M.; Jeřábek, K. Catalysis and polymer networks—The role of morphology and molecular accessibility. J. Mol. Catal. A Chem. 2001, 177, 3–20. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Hansen, T. Polymer-Supported Chiral Organocatalysts: Synthetic Strategies for the Road Towards Affordable Polymeric Immobilization. Eur. J. Org. Chem. 2010, 2010, 3179–3204. [Google Scholar] [CrossRef]
- Xu, W.; Xia, W.; Guan, Y.; Wang, Y.; Lu, C.; Yang, G.; Nie, J.; Chen, Z. DMAP-based flexible polymer networks formed via Heck coupling as efficient heterogeneous organocatalysts. React. Funct. Polym. 2016, 104, 15–21. [Google Scholar] [CrossRef]
- Schmiegel, C.J.; Baier, R.; Kuckling, D. Direct Asymmetric Aldol Reaction in Continuous Flow Using Gel-Bound Organocatalysts. Eur. J. Org. Chem. 2021, 2021, 2578–2586. [Google Scholar] [CrossRef]
- Kühbeck, D.; Saidulu, G.; Reddy, K.R.; Díaz, D.D. Critical assessment of the efficiency of chitosan biohydrogel beads as recyclable and heterogeneous organocatalyst for C–C bond formation. Green Chem. 2012, 14, 378–392. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kuckling, D.; Nebhani, L. Quinuclidine-Immobilized Porous Polymeric Microparticles as a Compelling Catalyst for the Baylis–Hillman Reaction. ACS Appl. Polym. Mater. 2022, 4, 8996–9005. [Google Scholar] [CrossRef]
- List, B.; Pojarliev, P.; Biller, W.T.; Martin, H.J. The proline-catalyzed direct asymmetric three-component Mannich reaction: Scope, optimization, and application to the highly enantioselective synthesis of 1,2-amino alcohols. J. Am. Chem. Soc. 2002, 124, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Llanes, P.; Rodríguez-Escrich, C.; Sayalero, S.; Pericàs, M.A. Organocatalytic Enantioselective Continuous-Flow Cyclopropanation. Org. Lett. 2016, 18, 6292–6295. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Mándity, I.M.; Fülöp, F. Highly selective deuteration of pharmaceutically relevant nitrogen-containing heterocycles: A flow chemistry approach. Mol. Divers. 2011, 15, 605–611. [Google Scholar] [CrossRef]
- Obst, F.; Simon, D.; Mehner, P.J.; Neubauer, J.W.; Beck, A.; Stroyuk, O.; Richter, A.; Voit, B.; Appelhans, D. One-step photostructuring of multiple hydrogel arrays for compartmentalized enzyme reactions in microfluidic devices. React. Chem. Eng. 2019, 4, 2141–2155. [Google Scholar] [CrossRef]
- Kleinschmidt, D.; Nothdurft, K.; Anakhov, M.V.; Meyer, A.A.; Mork, M.; Gumerov, R.A.; Potemkin, I.I.; Richtering, W.; Pich, A. Microgel organocatalysts: Modulation of reaction rates at liquid–liquid interfaces. Mater. Adv. 2020, 1, 2983–2993. [Google Scholar] [CrossRef]
- Henderson, W.A.; Schultz, C.J. The Nucleophilicity of Amines. J. Org. Chem. 1962, 27, 4643–4646. [Google Scholar] [CrossRef]
- Wu, L.; Yu, B.; Li, E.-Q. Recent Advances in Organocatalyst-Mediated Benzannulation Reactions. Adv. Synth. Catal. 2020, 362, 4010–4026. [Google Scholar] [CrossRef]
- Soleimani, E.; Khodaei, M.M.; Batooie, N.; Baghbanzadeh, M. Water-prompted synthesis of alkyl nitrile derivatives via Knoevenagel condensation and Michael addition reaction. Green Chem. 2011, 13, 566. [Google Scholar] [CrossRef]
- Xu, S.; Liao, Z.; Dianat, A.; Park, S.-W.; Addicoat, M.A.; Fu, Y.; Pastoetter, D.L.; Fabozzi, F.G.; Liu, Y.; Cuniberti, G.; et al. Combination of Knoevenagel Polycondensation and Water-Assisted Dynamic Michael-Addition-Elimination for the Synthesis of Vinylene-Linked 2D Covalent Organic Frameworks. Angew. Chem. Int. Ed Engl. 2022, 61, e202202492. [Google Scholar] [CrossRef] [PubMed]
- Majima, T.; Schnabel, W.; Weber, W. Phenyl-2,4,6-trimethylbenzoylphosphinates as water-soluble photoinitiators. Generation and reactivity of O=Ṗ(C6H5)(O−) radical anions. Makromol. Chem. 1991, 192, 2307–2315. [Google Scholar] [CrossRef]
- Simon, D.; Obst, F.; Haefner, S.; Heroldt, T.; Peiter, M.; Simon, F.; Richter, A.; Voit, B.; Appelhans, D. Hydrogel/enzyme dots as adaptable tool for non-compartmentalized multi-enzymatic reactions in microfluidic devices. React. Chem. Eng. 2019, 4, 67–77. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition Code | Catalytic Monomer | Complimentary Monomer | Crosslinker | UV Irradiation Time (s) | UV Intensity (W) | Number of Gel Dots |
|---|---|---|---|---|---|---|
| A | DMAPAM | DMAM | MBMA | 8 | 0.42 | 202 |
| B | 5 | MMA | EGDMA | 14 | 0.42 | 202 |
| C | 4 | DMAM | MBMA | 90 | 1.28 | 202 |
| Composition Code | Solvent Uptake (%) | Volume Degree of Swelling (mm2) | ||
|---|---|---|---|---|
| * DMSO:IP | * DMSO:IP:W | * DMSO:IP | * DMSO:IP:W | |
| A | 266 ± 0.1 | 211 ± 14 | 2.25 | 1.85 |
| B | 220 ± 21 | 165 ± 18 | 2.10 | 2.08 |
| C | 23 ± 5 | 177 ± 19 | 1.00 | 1.61 |
| Entry | Reactant 1 | Reactant 2 | Solvent Mixture | Conversion (%) with Different Compositions | ||
|---|---|---|---|---|---|---|
| Gel Dot Composition A | Gel Dot Composition B | Gel Dot Composition C | ||||
| 1–3 |  MBA |  MN | * DMSO:IP | 77 | 87 | 67 |
| 4–6 | MBA | MN | * DMSO:IP:W | 93 | 100 | 98 |
| 7–9 |  BA | MN | * DMSO:IP | 38 | 99 | 82 |
| 10–12 | BA | MN | * DMSO:IP:W | 81 | 86 | 98 |
| 13–15 |  IBA | MN | * DMSO:IP | 59 | 75 | 54 |
| 16–18 | IBA | MN | * DMSO:IP:W | 52 | 83 | 91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Killi, N.; Bartenbach, J.; Kuckling, D. Polymeric Networks Containing Amine Derivatives as Organocatalysts for Knoevenagel Reaction within Continuously Driven Microfluidic Reactors. Gels 2023, 9, 171. https://doi.org/10.3390/gels9030171
Killi N, Bartenbach J, Kuckling D. Polymeric Networks Containing Amine Derivatives as Organocatalysts for Knoevenagel Reaction within Continuously Driven Microfluidic Reactors. Gels. 2023; 9(3):171. https://doi.org/10.3390/gels9030171
Chicago/Turabian StyleKilli, Naresh, Julian Bartenbach, and Dirk Kuckling. 2023. "Polymeric Networks Containing Amine Derivatives as Organocatalysts for Knoevenagel Reaction within Continuously Driven Microfluidic Reactors" Gels 9, no. 3: 171. https://doi.org/10.3390/gels9030171
APA StyleKilli, N., Bartenbach, J., & Kuckling, D. (2023). Polymeric Networks Containing Amine Derivatives as Organocatalysts for Knoevenagel Reaction within Continuously Driven Microfluidic Reactors. Gels, 9(3), 171. https://doi.org/10.3390/gels9030171