Roles of Non-Coding RNA in Sugarcane-Microbe Interaction

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
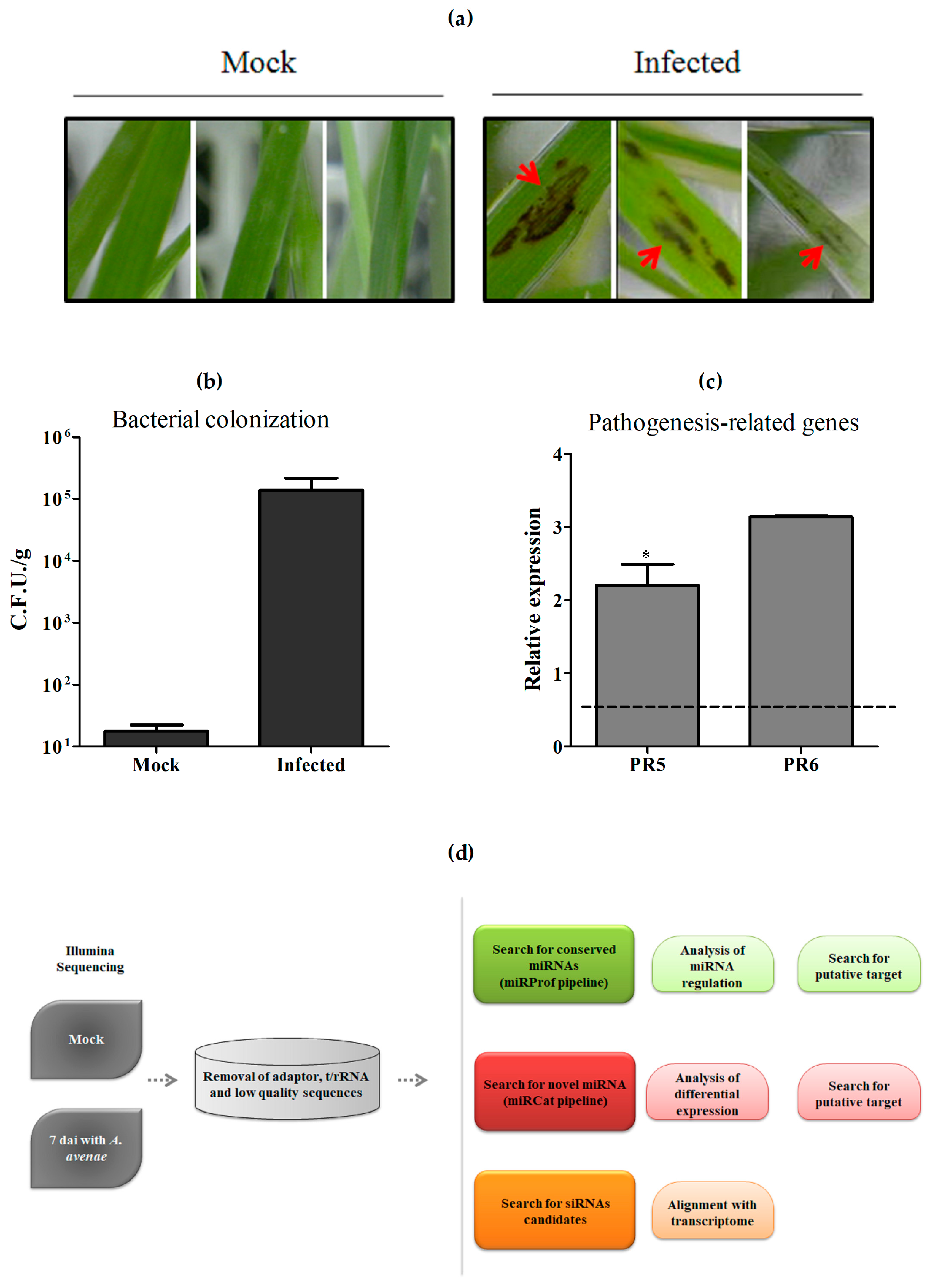
2.1. Pathogen Assay and Small RNA Sequencing
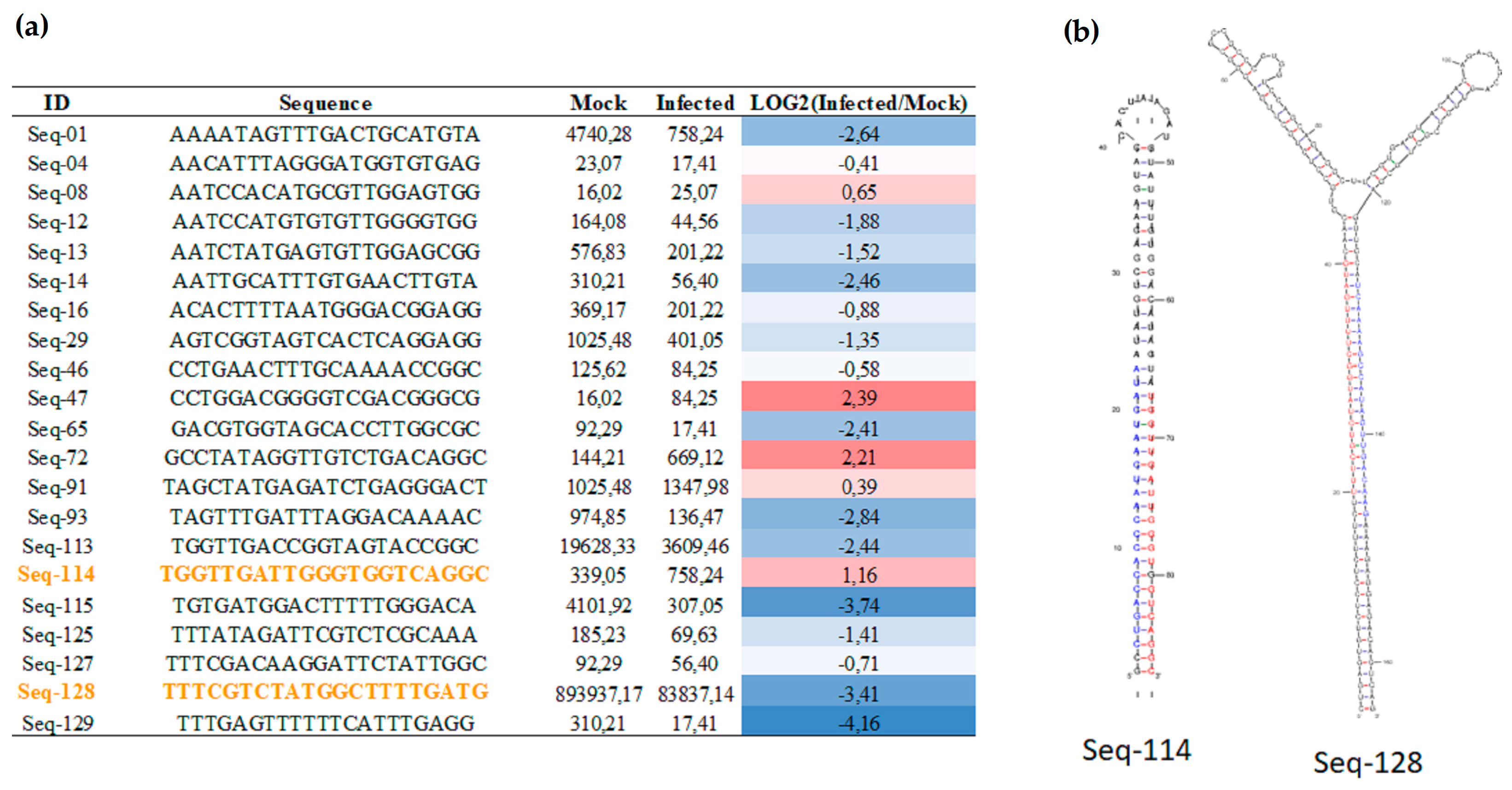
2.2. Sugarcane lincRNAs
2.3. Novel Sugarcane miRNAs Were Identified
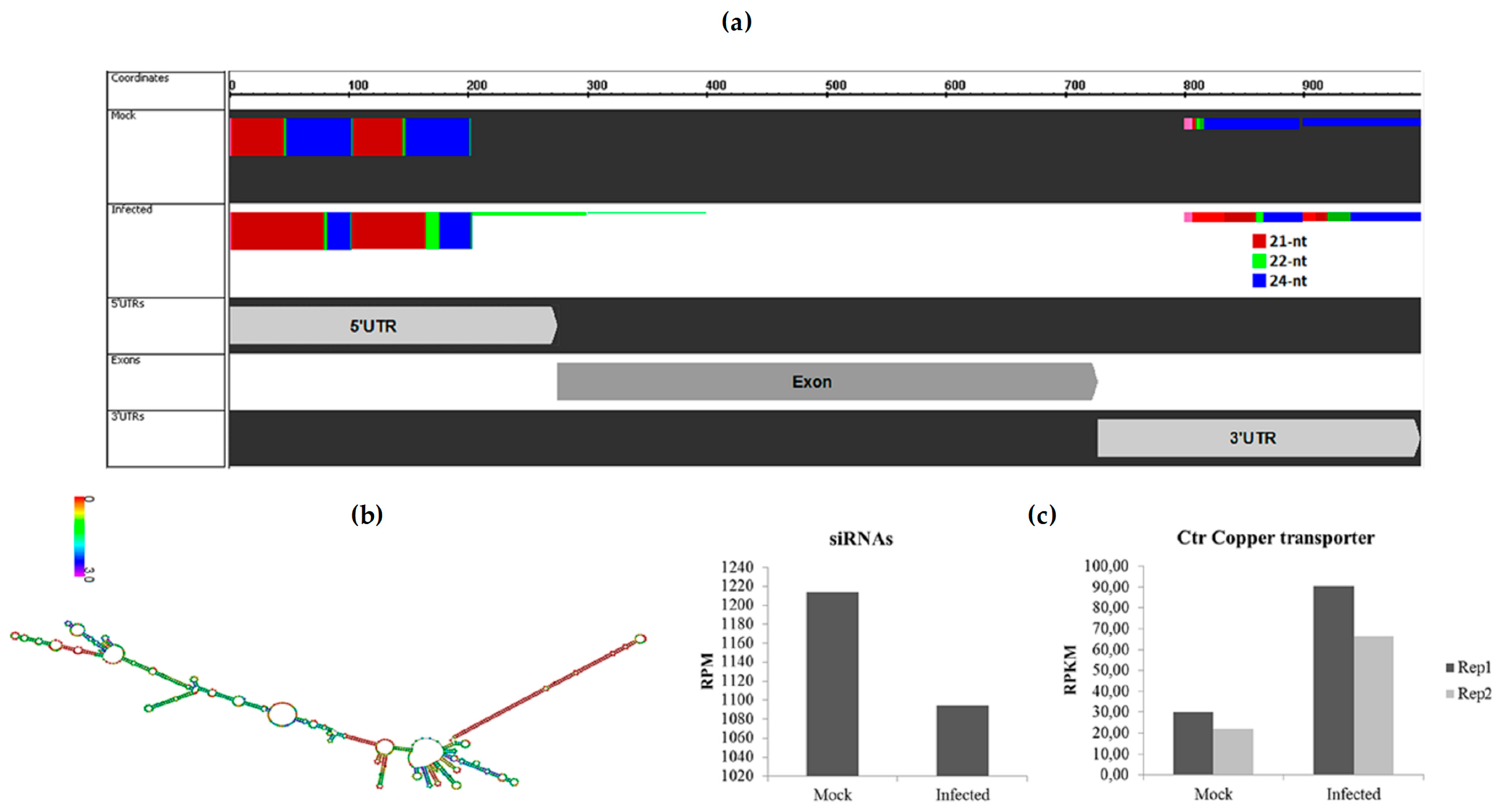
2.4. Sugarcane siRNA Were Identified
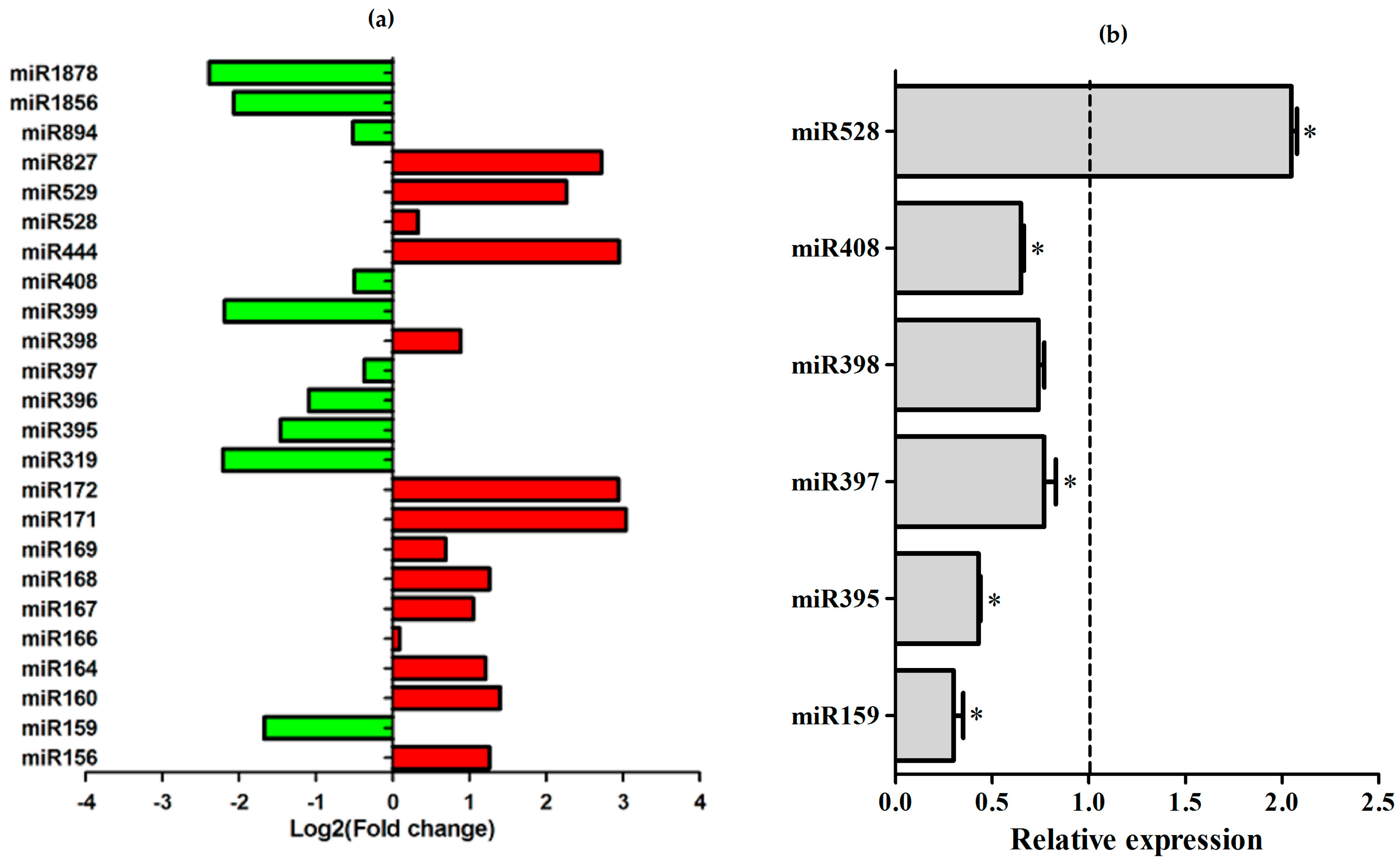
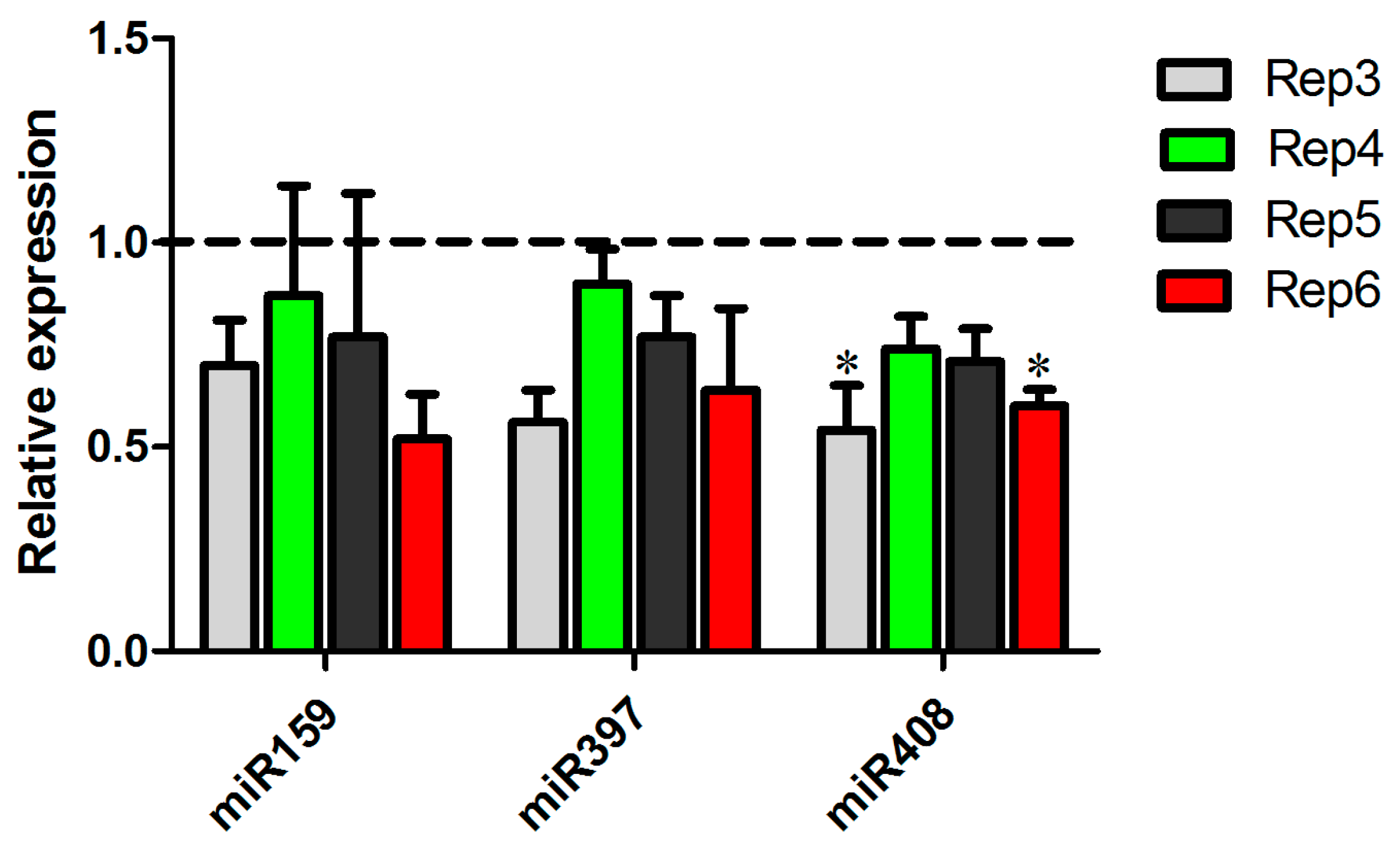
2.5. Differential Expression of Conserved Sugarcane miRNAs in Response to A. avenae Infection
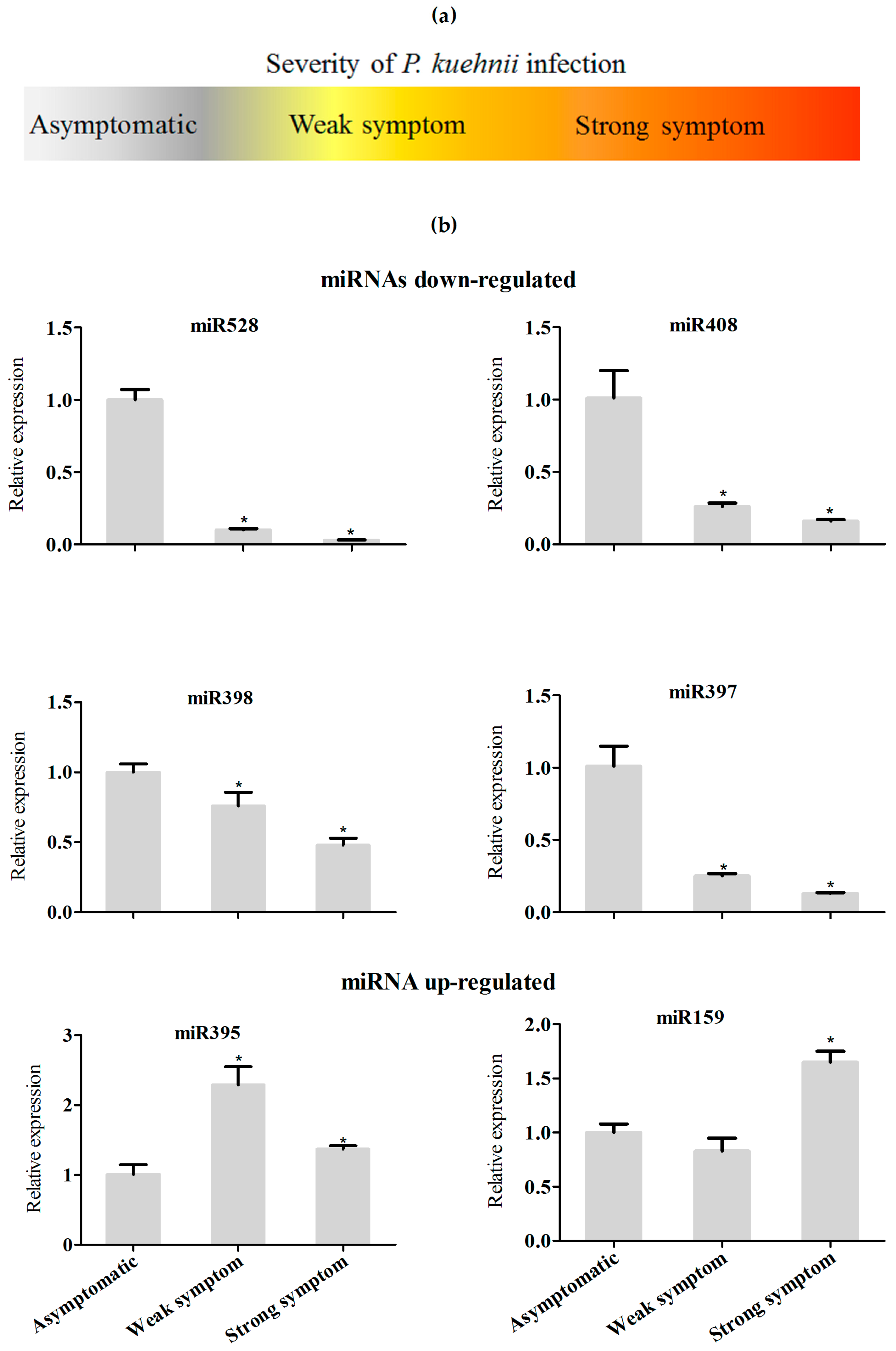
2.6. microRNAs Exhibiting Similar Regulation in Response to Different Pathogens
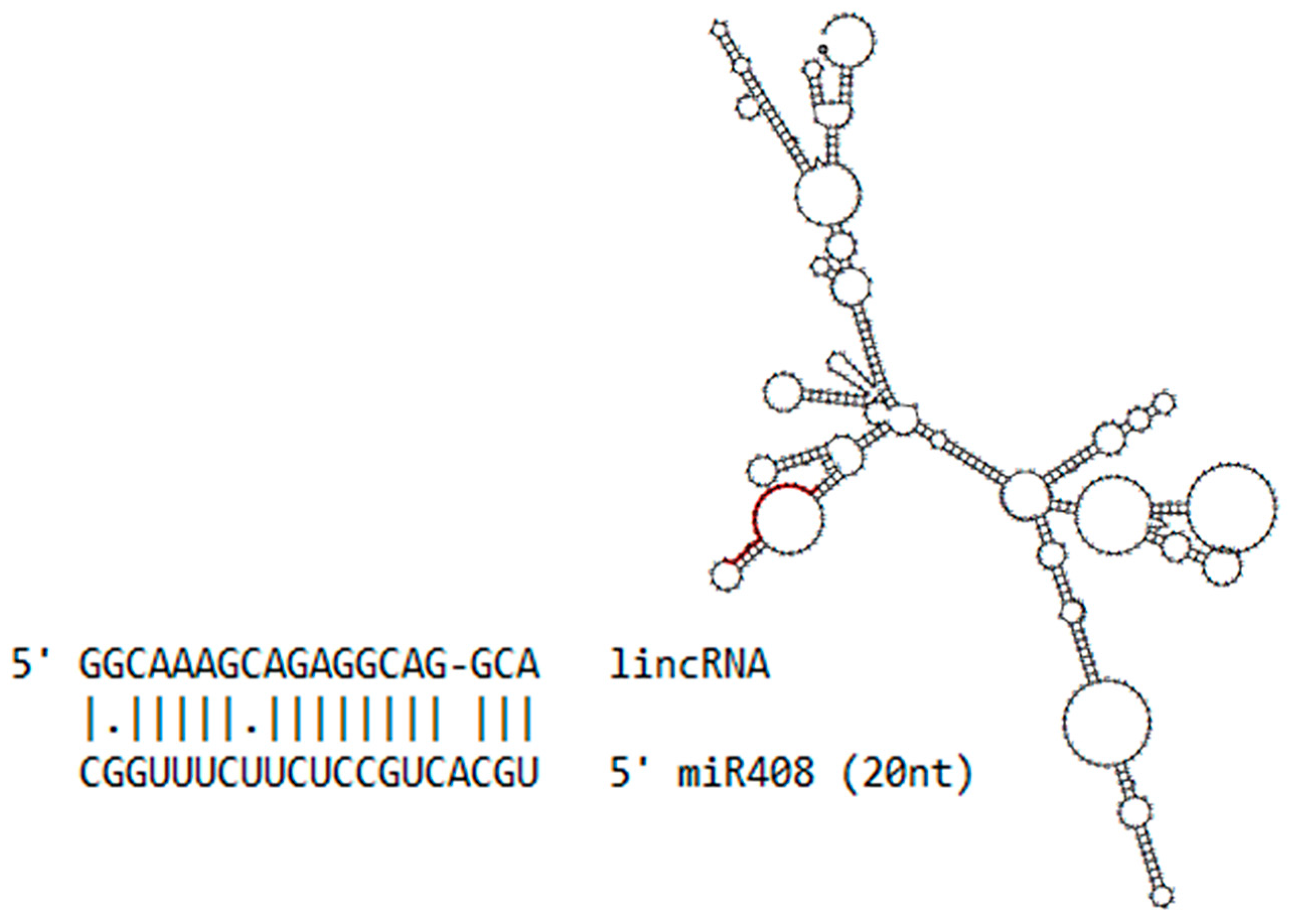
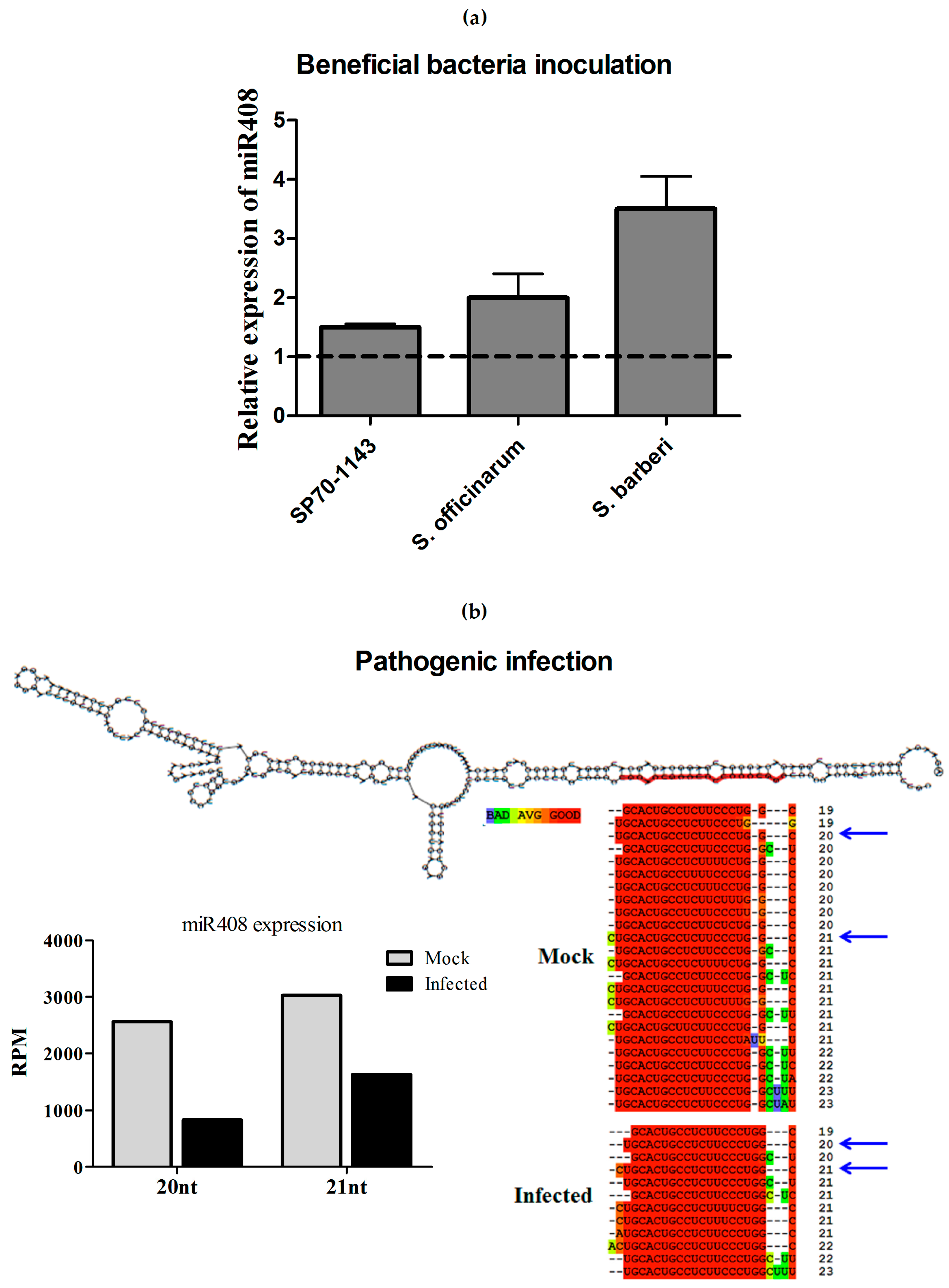
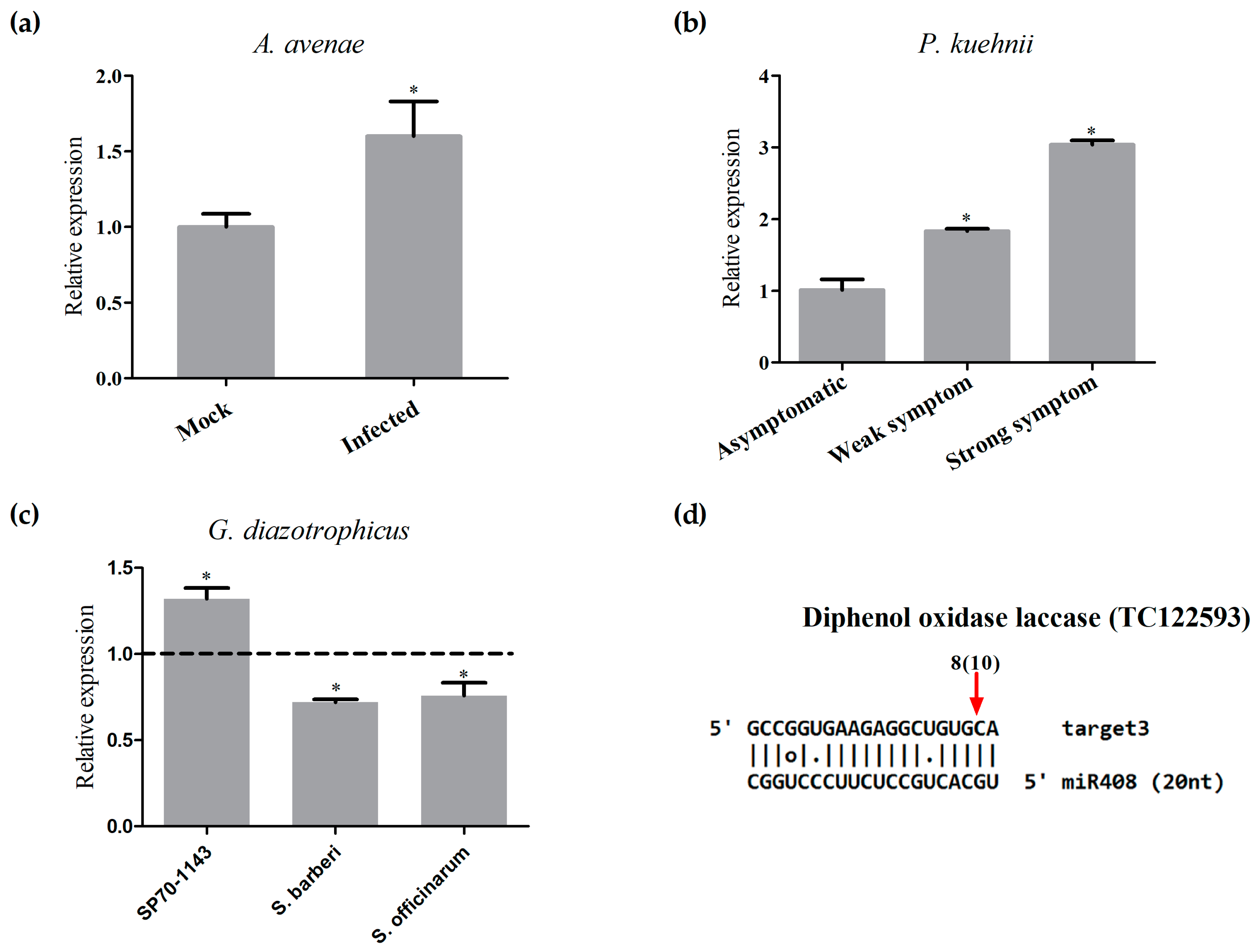
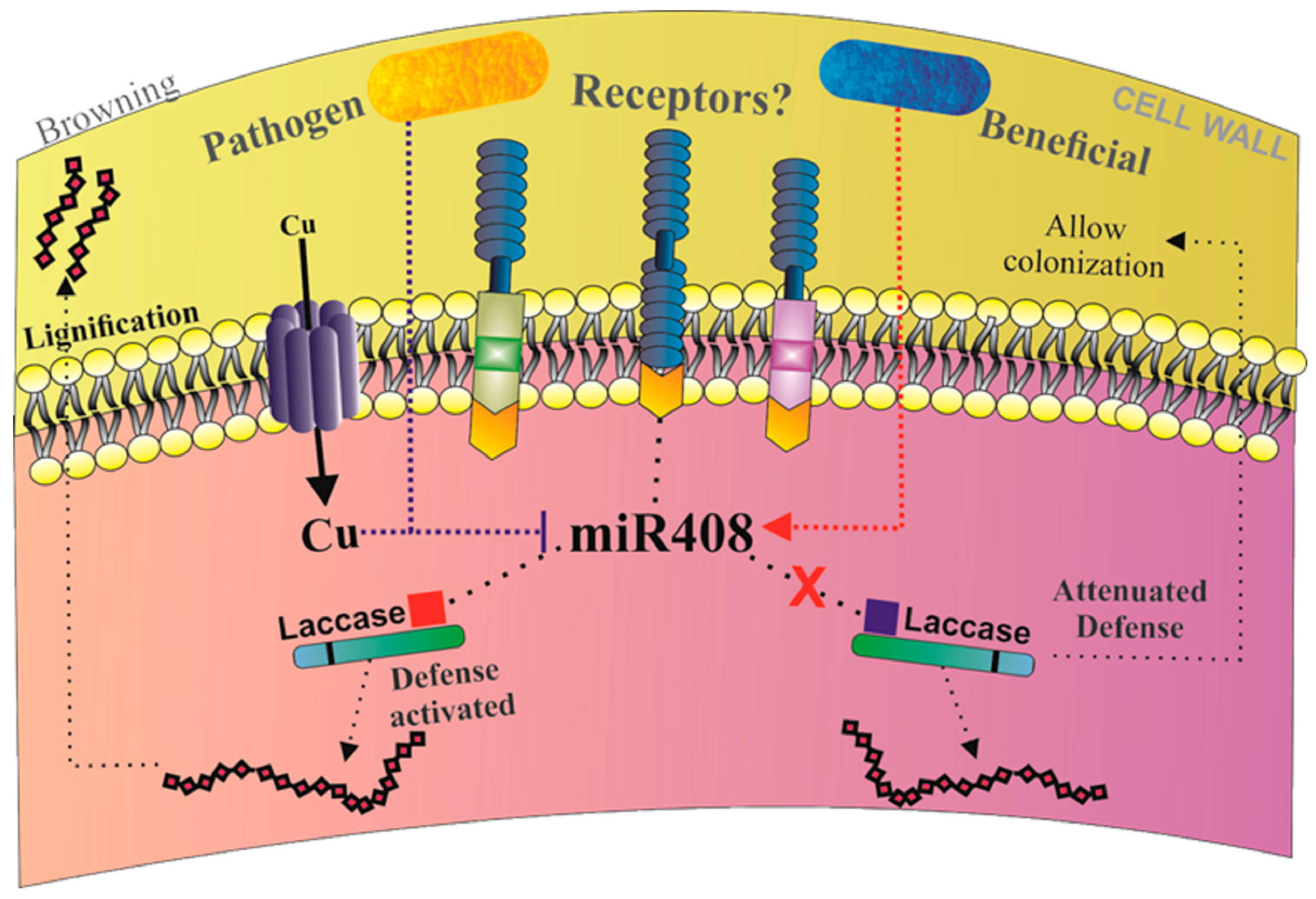
2.7. Differential Regulation of miR408 in Sugarcane Infected with Pathogens or Beneficial Bacteria
3. Discussion
4. Materials and Methods
4.1. Pathogen Infection Assay
4.2. Validation of A. avenae Infection
4.3. Treatments with Beneficial Diazotrophic Bacteria
4.4. RNA Extraction and Sequencing Small RNA Library Construction
4.5. Bioinformatics Analysis
4.5.1. Identification of sRNAs
4.5.2. Identification of lincRNA
4.6. Validation of miRNA and Target Gene Expression by qRT-PCR
4.7. Semi-Quantitative RT-PCR
4.8. Modified 5′ RACE Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Azevedo, R.A.; Carvalho, R.F.; Cia, M.C.; Gratão, P.L. Sugarcane Under Pressure: An Overview of Biochemical and Physiological Studies of Abiotic Stress. Trop. Plant Biol. 2011, 4, 42–51. [Google Scholar] [CrossRef]
- Grivet, L.; Arruda, P. Sugarcane genomics: depicting the complex genome of an important tropical crop. Curr. Opin. Plant Biol. 2002, 5, 122–127. [Google Scholar] [CrossRef]
- Jannoo, N.; Grivet, L.; Chantret, N.; Garsmeur, O.; Glaszmann, J.; Arruda, P.; D’Hont, A. Orthologous comparison in a gene-rich region among grasses reveals stability in the sugarcane polyploid genome. Plant J. 2007, 50, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Rott, P.; Bailey, A.R.; Comstock, J.C.; Croft, B.J.; Saumtally, A.S. Diseases caused by bacteria. In A Guide to Sugarcane Diseases; Editions Quae: Versailles, France, 2000; pp. 21–67. [Google Scholar]
- Chartered Management Institute (CMI). Acidovorax avenae subsp. avenae. In Distribution Maps of Plant Diseases; CMI: London, UK, 1995; p. 511. [Google Scholar]
- Willems, A.; Goo, M.; Thielemans, S.; Gillis, M.; Kersters, K.; Ley, D.E. Transfer of Several Phytopathogenic Pseudomonas Species to Acidovorax as Acidovorax avenae subsp. avenae subsp. nov., comb. nov., Acidovorax avenae subsp. citrulli, Acidovorax avenae subsp. cattleyae, and Acidovorax konjaci. Int. J. Syst. Bacteriol. 1992, 42, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Fegan, M. Plant pathogenic members of the genera ACIDOVORAX and HERBASPIRILLUM. Plant-Assoc. Bact. 2006, 671–702. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational Identification of Plant MicroRNAs and Their Targets, Including a Stress-Induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J. Novel and Stress-Regulated MicroRNAs and Other Small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Brtela, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Llave, C.; Kasschau, K.D.; Rector, M.A.; Carrington, J.C. Endogenous and Silencing-Associated Small RNAs in Plants. Society 2002, 14, 1605–1619. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and Comparison of Small RNAs from Plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Kidner, C.A.; Martienssen, R.A. The developmental role of microRNA in plants. Curr. Opin. Plant Biol. 2005, 8, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D.G. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Katiyar-agarwal, S.; Gao, S.; Vivian-smith, A.; Jin, H. A novel class of bacteria-induced small RNAs in Arabidopsis. Genes Dev. 2007, 21, 3123–3134. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Fu, Y.; Sunkar, R.; Barbazuk, W.B.; Zhu, J.-K.; Yu, O. Novel and nodulation-regulated microRNAs in soybean roots. BMC Genom. 2008, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Soto-Suárez, M.; Baldrich, P.; Weigel, D.; Rubio-Somoza, I.; San Segundo, B. The Arabidopsis miR396 mediates pathogen-associated molecular pattern-triggered immune responses against fungal pathogens. Sci. Rep. 2017, 7, 44898. [Google Scholar] [CrossRef] [PubMed]
- Zogli, P.; Libault, M. Plant response to biotic stress: Is there a common epigenetic response during plant-pathogenic and symbiotic interactions? Plant Sci. 2017, 263, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Wang, M.B. Molecular functions of long non-coding RNAs in plants. Genes (Basel) 2012, 3, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, L.; Li, D.; Zhu, L.; Hu, S. Long Non-coding RNAs and Their Biological Roles in Plants. Genom. Proteom. Bioinform. 2015, 13, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y. Biochemical and Biophysical Research Communications Long noncoding RNAs: New regulators in plant development. Biochem. Biophys. Res. Commun. 2013, 436, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Stephen, S.; Taylor, J.; Helliwell, C.A.; Wang, M.B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014, 201, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Han, Z.; Guo, Q.; Liu, Y.; Zheng, Y.; Wu, F.; Jin, W. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Pilon, M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem. 2008, 283, 15932–15945. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-F.; Zhu, C. The role of microRNAs in copper and cadmium homeostasis. Biochem. Biophys. Res. Commun. 2009, 386, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Burkhead, J.L.; Reynolds, K.A.G.; Abdel-Ghany, S.E.; Cohu, C.M.; Pilon, M. Copper homeostasis. New Phytol. 2009, 182, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Sato, F. Plant pathogenesis-related proteins: molecular mechanisms of gene expression and protein function. J. Biochem. 1999, 125, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Santa Brigida, A.B.; Rojas, C.A.; Grativol, C.; De Armas, E.M.; Entenza, J.O.P.; Thiebaut, F.; Lima, M.D.F.; Farrinelli, L.; Hemerly, A.S.; et al. Sugarcane transcriptome analysis in response to infection caused by Acidovorax avenae subsp. avenae. PLoS ONE 2016, 11, e0166473. [Google Scholar] [CrossRef] [PubMed]
- Stocks, M.B.; Moxon, S.; Mapleson, D.; Woolfenden, H.C.; Mohorianu, I.; Folkes, L.; Schwach, F.; Dalmay, T.; Moulton, V. The UEA sRNA workbench: A suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 2012, 28, 2059–2061. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.; Grativol, C.; Thiebaut, F.; Carvalho, T.; Hardoim, P.; Hemerly, A.; Lifschitz, S.; Ferreira, P.; Walter, M. PlantRNA_Sniffer: A SVM-Based Workflow to Predict Long Intergenic Non-Coding RNAs in Plants. Non-Coding RNA 2017, 3, 11. [Google Scholar] [CrossRef]
- Thiebaut, F.; Grativol, C.; Carnavale-bottino, M.; Rojas, C.A.; Farinelli, L.; Hemerly, A.S.; Tanurdzic, M.; Martienssen, R.A.; Ferreira, P.C.G. Computational identification and analysis of novel sugarcane microRNAs. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Grativol, C.; Regulski, M.; Bertalan, M.; McCombie, W.R.; Da Silva, F.R.; Zerlotini Neto, A.; Vicentini, R.; Farinelli, L.; Hemerly, A.S.; Martienssen, R.A.; et al. Sugarcane genome sequencing by methylation filtration provides tools for genomic research in the genus Saccharum. Plant J. 2014, 79, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Institute for Theoretical Chemistry RNAfold WebServer. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RN (accessed on 19 December 2017).
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Glynn, N.C.; Glaz, B.; Comstock, J.C.; Sood, S.; Station, S.F. Orange Rust Effects on Leaf Photosynthesis and Related Characters of Sugarcane. Plant Dis. 2011, 95, 640–647. [Google Scholar] [CrossRef]
- De Souza, R.S.C.; Okura, V.K.; Armanhi, J.S.L.; Jorrín, B.; Lozano, N.; da Silva, M.J.; González-Guerrero, M.; de Araújo, L.M.; Ferreira, N.V.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Nat. Sci. Rep. 2016, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maunoury, N.; Vaucheret, H. AGO1 and AGO2 act redundantly in miR408-mediated Plantacyanin regulation. PLoS ONE 2011, 6, e28729. [Google Scholar] [CrossRef] [PubMed]
- Katiyar-Agarwal, S.; Jin, H. Role of small RNAs in host-microbe interactions. Annu. Rev. Phytopathol. 2010, 48, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ferrer, V.; Voinnet, O. Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 2009, 60, 485–510. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.D.; Rago, A.M.; Fontana, C.A.; Vignolo, G.M.; Cocconcelli, P.S.; Mariotti, J.A. Isolation and genetic characterization of Acidovorax avenae from red stripe infected sugarcane in Northwestern Argentina. Eur. J. Plant Pathol. 2013, 137, 525–534. [Google Scholar] [CrossRef]
- Christopher, W.N.; Edgerton, C.W. Bacterial stripe diseases of sugar cane in Louisiana. J. Agric. Res. 1932, 41, 259–267. [Google Scholar]
- Hayward, A.C. Studies on bacterial pathogens of sugar cane. II. Differentiation, taxonomy and nomenclature of the bacteria causing red stripe and mottled stripe diseases. Mauritius Sugar Ind. Res. Inst. Occas. Pap. 1962, 13, 13–17. [Google Scholar]
- Shakya, D.D.; Chung, H.S.; Vinther, F. Transmission of Pseudomonas avenae, the cause of bacterial stripe of rice. J. Phytopathol. Z. 1986, 116, 92–96. [Google Scholar] [CrossRef]
- Paicu, C.; Mohorianu, I.; Stocks, M.; Xu, P.; Coince, A.; Billmeier, M.; Dalmay, T.; Moulton, V.; Moxon, S. miRCat2: Accurate prediction of plant and animal microRNAs from next-generation sequencing datasets. Bioinformatics 2017, 33, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Lelandais-Brière, C.; Naya, L.; Sallet, E.; Calenge, F.; Frugier, F.; Hartmann, C.; Gouzy, J.; Crespi, M. Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell 2009, 21, 2780–2796. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Chu, Z.; Li, X.; Xu, C.; Wang, S. The bacterial pathogen Xanthomonas oryzae overcomes rice defenses by regulating host copper redistribution. Plant Cell 2010, 22, 3164–3176. [Google Scholar] [CrossRef] [PubMed]
- Kasschau, K.D.; Fahlgren, N.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Carrington, J.C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007, 5, e57. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.; Kanno, T.; Huettel, B.; Daxinger, L.; Matzke, A.J.M. Targets of RNA-directed DNA methylation. Curr. Opin. Plant Biol. 2007, 10, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Sancenón, V.; Puig, S.; Mateu-Andrés, I.; Dorcey, E.; Thiele, D.J.; Peñarrubia, L. The Arabidopsis Copper Transporter COPT1 Functions in Root Elongation and Pollen Development. J. Biol. Chem. 2004, 279, 15348–15355. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Abdel-Ghany, S.E.; Cohu, C.M.; Kobayashi, Y.; Shikanai, T.; Pilon, M. Regulation of copper homeostasis by micro-RNA in Arabidopsis. J. Biol. Chem. 2007, 282, 16369–16378. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lu, Z. The fate of miRNA* strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS ONE 2010, 5, e11387. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, S.; Zhou, X.; Chellappan, P.; Chen, Z.; Zhou, X.; Zhang, X.; Fromuth, N.; Coutino, G.; Coffey, M.; et al. Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol. Biol. 2011, 75, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Hao, Z.; Yan, J.; Li, G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genom. 2015, 16, 793. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.A.; Paz-Ares, J.; et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Saharan, B.; Nehra, V. Plant Growth Promoting Rhizobacteria: a critical review. Life Sci. Med. Res. 2011, 2011, 1–30. [Google Scholar]
- James, E.K.; Olivares, F.L.; de Oliveira, A.L.; dos Reis, F.B.; da Silva, L.G.; Reis, V.M. Further observations on the interaction between sugar cane and Gluconacetobacter diazotrophicus under laboratory and greenhouse conditions. J. Exp. Bot. 2001, 52, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Döbereiner, J.; Baldani, V.L.D.; Reis, V.M. Endophytic Occurrence of Diazotrophic Bacteria in Non-Leguminous Crops. In Azospirillum VI and Related Microorganisms; Springer: Berlin/Heidelberg, Germany, 1995; pp. 3–14. [Google Scholar]
- Vinagre, F.; Vargas, C.; Schwarcz, K.; Cavalcante, J.; Nogueira, E.M.; Baldani, J.I.; Ferreira, P.C.G.; Hemerly, A.S. SHR5: A novel plant receptor kinase involved in plant-N2-fixing endophytic bacteria association. J. Exp. Bot. 2006, 57, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, J.J.V.; Vargas, C.; Nogueira, E.M.; Vinagre, F.; Schwarcz, K.; Baldani, J.I. Members of the ethylene signalling pathway are regulated in sugarcane during the association with nitrogen-fixing endophytic bacteria. J. Exp. Bot. 2007, 58, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Branscheid, A.; Sieh, D.; Pant, B.D.; May, P.; Devers, E.A.; Elkrog, A.; Schauser, L.; Scheible, W.-R.; Krajinski, F. Expression pattern suggests a role of MiR399 in the regulation of the cellular response to local Pi increase during arbuscular mycorrhizal symbiosis. Mol. Plant-Microbe. Interact. 2010, 23, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Jay, F.; Nomura, K.; He, S.Y.; Voinnet, O. Suppression of the microRNA pathway by bacterial effector proteins. Science 2008, 321, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Quintero, A.L.; Sablok, G.; Tatarinova, T.V.; Conesa, A.; Kuo, J.; López, C. Mining of miRNAs and potential targets from gene oriented clusters of transcripts sequences of the anti-malarial plant, Artemisia annua. Biotechnol. Lett. 2012, 34, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Grativol, C.; Hemerly, A.S.; Ferreira, P.C.G. MicroRNA Networks in Plant-Microorganism Interactions. Trop. Plant Biol. 2015, 8, 40–50. [Google Scholar] [CrossRef]
- Carvalho, T.L.G.; Ballesteros, H.G.F.; Thiebaut, F.; Ferreira, P.C.G.; Hemerly, A.S. Nice to meet you: genetic, epigenetic and metabolic controls of plant perception of beneficial associative and endophytic diazotrophic bacteria in non-leguminous plants. Plant Mol. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Rojas, C.A.; Grativol, C.; Motta, M.; Vieira, T.; Regulski, M.; Martienssen, R.A.; Farinelli, L.; Hemerly, A.S.; Ferreira, P.C. Genome-wide identification of microRNA and siRNA responsive to endophytic beneficial diazotrophic bacteria in maize. BMC Genom. 2014, 15, 766. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-H.; Fan, L.; Liu, Y.; Xu, H.; Llewellyn, D.; Wilson, I. miR482 regulation of NBS-LRR defense genes during fungal pathogen infection in cotton. PLoS ONE 2013, 8, e84390. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Zhang, J.; Wu, L.; Qi, Y.; Zhou, J.-M. Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol. 2010, 152, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, D.M.; Whetten, R.; Bao, W.; Chen, C.-L.; Sederoff, R.R. The role of of laccase in lignification. Plant J. 1993, 4, 751–757. [Google Scholar] [CrossRef]
- Dean, J.F.D.; Eriksson, K.-E.L. Laccase and the Deposition of Lignin in Vascular Plants. Holzforsch. Int. J. Biol. Chem. Phys. Technol. Wood 1994, 48, 21–33. [Google Scholar] [CrossRef]
- Lamb, C.; Dixon, R. The oxidative burst in plant disease resistance. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 251–275. [Google Scholar] [CrossRef] [PubMed]
- Mithöfer, A.; Schulze, B.; Boland, W. Biotic and heavy metal stress response in plants: evidence for common signals. FEBS Lett. 2004, 566, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Yang, C.; Chiang, V.L. Conservation and diversity of microRNA-associated copper-regulatory networks in Populus trichocarpa. J. Integr. Plant Biol. 2011, 53, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Whetten, R.; Sederoff, R. Lignin Biosynthesis. Plant Cell 1995, 7, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Constabel, C.; Ryan, C. A survey of wound- and methyl jasmonate-induced leaf polyphenol oxidase in crop plants. Phytochemistry 1998, 47, 507–511. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Dewey, R.E.; Boss, W.; Phillippy, B.Q.; Qu, R. Enhanced Agrobacterium-mediated transformation efficiencies in monocot cells is associated with attenuated defense responses. Plant Mol. Biol. 2013, 81, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Reis, V.M.; Olivares, F.L.F.; Döbereiner, J. Improved methodology for isolation of Acetobacter diazotrophicus and confirmation of its endophytic habitat. World J. Microbiol. Biotechnol. 1994, 10, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Vargas, L.; Carvalho, T.L.G.; Ferreira, P.C.G.; Baldani, V.L.D.; Baldani, J.I.; Hemerly, A.S. Early responses of rice (Oryza sativa L.) seedlings to inoculation with beneficial diazotrophic bacteria are dependent on plant and bacterial genotypes. Plant Soil 2012, 356, 127–137. [Google Scholar] [CrossRef]
- Thiebaut, F.; Rojas, C.A.; Almeida, K.L.; Grativol, C.; Domiciano, G.C.; Lamb, C.R.C.; De Almeida Engler, J.; Hemerly, A.S.; Ferreira, P.C.G. Regulation of miR319 during cold stress in sugarcane. Plant Cell Environ. 2012, 35, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Zanca, A.S.; Vicentini, R.; Ortiz-Morea, F.A.; Del Bem, L.E.V.; da Silva, M.J.; Vincentz, M.; Nogueira, F.T.S. Identification and expression analysis of microRNAs and targets in the biofuel crop sugarcane. BMC Plant Biol. 2010, 10, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoagland, D.; Arnon, D. The Water-Culture Method for Growing Plants Without Soil, 347th ed.; College of Agriculture, University of California: Berkeley, CA, USA, 1950. [Google Scholar]
- Vargas, L.; Brigida, A.B.S.; Mota Filho, J.P.; De Carvalho, T.G.; Rojas, C.A.; Vaneechoutte, D.; Van Bel, M.; Farrinelli, L.; Ferreira, P.C.G.; Vandepoele, K.; et al. Drought tolerance conferred to sugarcane by association with gluconacetobacter diazotrophicus: A transcriptomic view of hormone pathways. PLoS ONE 2014, 9, e114744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCBI Gene Expression Omnibus.
- Moxon, S.; Jing, R.; Szittya, G.; Schwach, F.; Rusholme Pilcher, R.L.; Moulton, V.; Dalmay, T. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; Van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, H.; Sharma, C.; Khaitovich, P.; Vogel, J.; Stadler, P.; Hackermueller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | Replicate 1 | |
|---|---|---|
| Mock | Infected | |
| All reads | 2,209,310 | 2,852,027 |
| t/rRNA filtering 1 | 1,714,978 | 1,668,744 |
| Low quality reads filtering 2 | 1,560,245 | 1,436,225 |
| Conserved miRNAs 3 | 105,279 | 82,728 |
| Unique | ||
| t/rRNA filtering 1 | 930,266 | 802,581 |
| Low quality reads 2 | 912,871 | 780,141 |
| Conserved miRNAs 3 | 1872 | 2313 |
| Novel miRNAs 4 | 86 | 67 |
| Putative siRNA 5 | 910,913 | 777,761 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiebaut, F.; Rojas, C.A.; Grativol, C.; Calixto, E.P.d.R.; Motta, M.R.; Ballesteros, H.G.F.; Peixoto, B.; De Lima, B.N.S.; Vieira, L.M.; Walter, M.E.; et al. Roles of Non-Coding RNA in Sugarcane-Microbe Interaction. Non-Coding RNA 2017, 3, 25. https://doi.org/10.3390/ncrna3040025
Thiebaut F, Rojas CA, Grativol C, Calixto EPdR, Motta MR, Ballesteros HGF, Peixoto B, De Lima BNS, Vieira LM, Walter ME, et al. Roles of Non-Coding RNA in Sugarcane-Microbe Interaction. Non-Coding RNA. 2017; 3(4):25. https://doi.org/10.3390/ncrna3040025
Chicago/Turabian StyleThiebaut, Flávia, Cristian A. Rojas, Clícia Grativol, Edmundo P. da R. Calixto, Mariana R. Motta, Helkin G. F. Ballesteros, Barbara Peixoto, Berenice N. S. De Lima, Lucas M. Vieira, Maria Emilia Walter, and et al. 2017. "Roles of Non-Coding RNA in Sugarcane-Microbe Interaction" Non-Coding RNA 3, no. 4: 25. https://doi.org/10.3390/ncrna3040025
APA StyleThiebaut, F., Rojas, C. A., Grativol, C., Calixto, E. P. d. R., Motta, M. R., Ballesteros, H. G. F., Peixoto, B., De Lima, B. N. S., Vieira, L. M., Walter, M. E., De Armas, E. M., Entenza, J. O. P., Lifschitz, S., Farinelli, L., Hemerly, A. S., & Ferreira, P. C. G. (2017). Roles of Non-Coding RNA in Sugarcane-Microbe Interaction. Non-Coding RNA, 3(4), 25. https://doi.org/10.3390/ncrna3040025