Dynamic Expression of Long Non-Coding RNAs Throughout Parasite Sexual and Neural Maturation in Schistosoma Japonicum


Abstract
:1. Introduction
2. Results
2.1. Thousands of lncRNAs are Expressed in S. japonicum
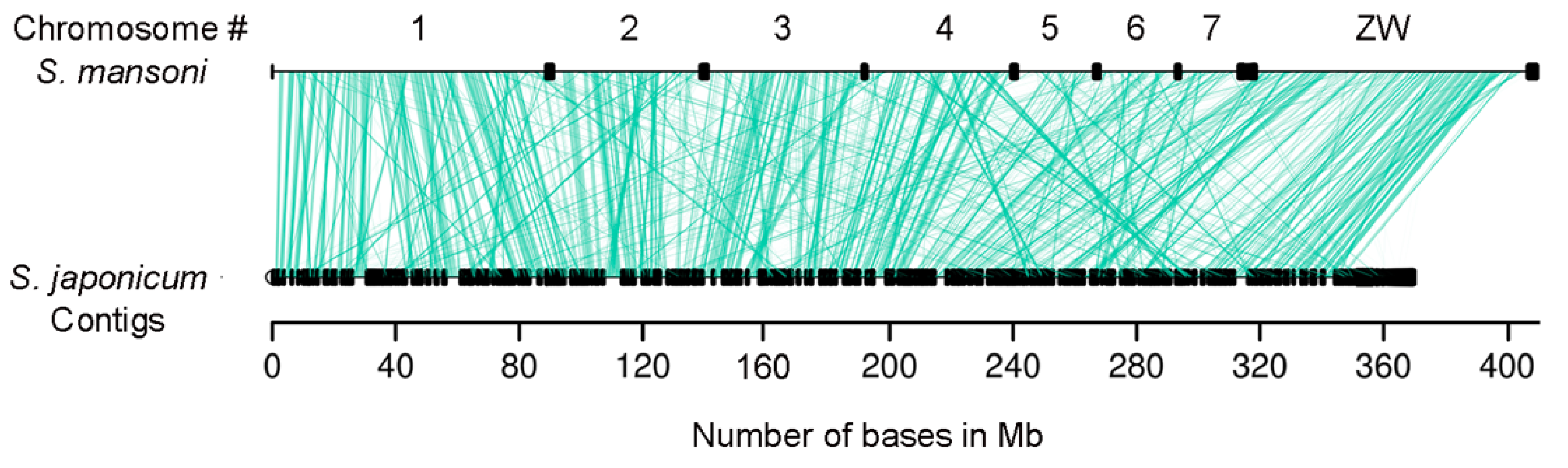
2.2. Synteny Conservation is Higher than Gene Sequence Similarity in Schistosoma lncRNA Genes
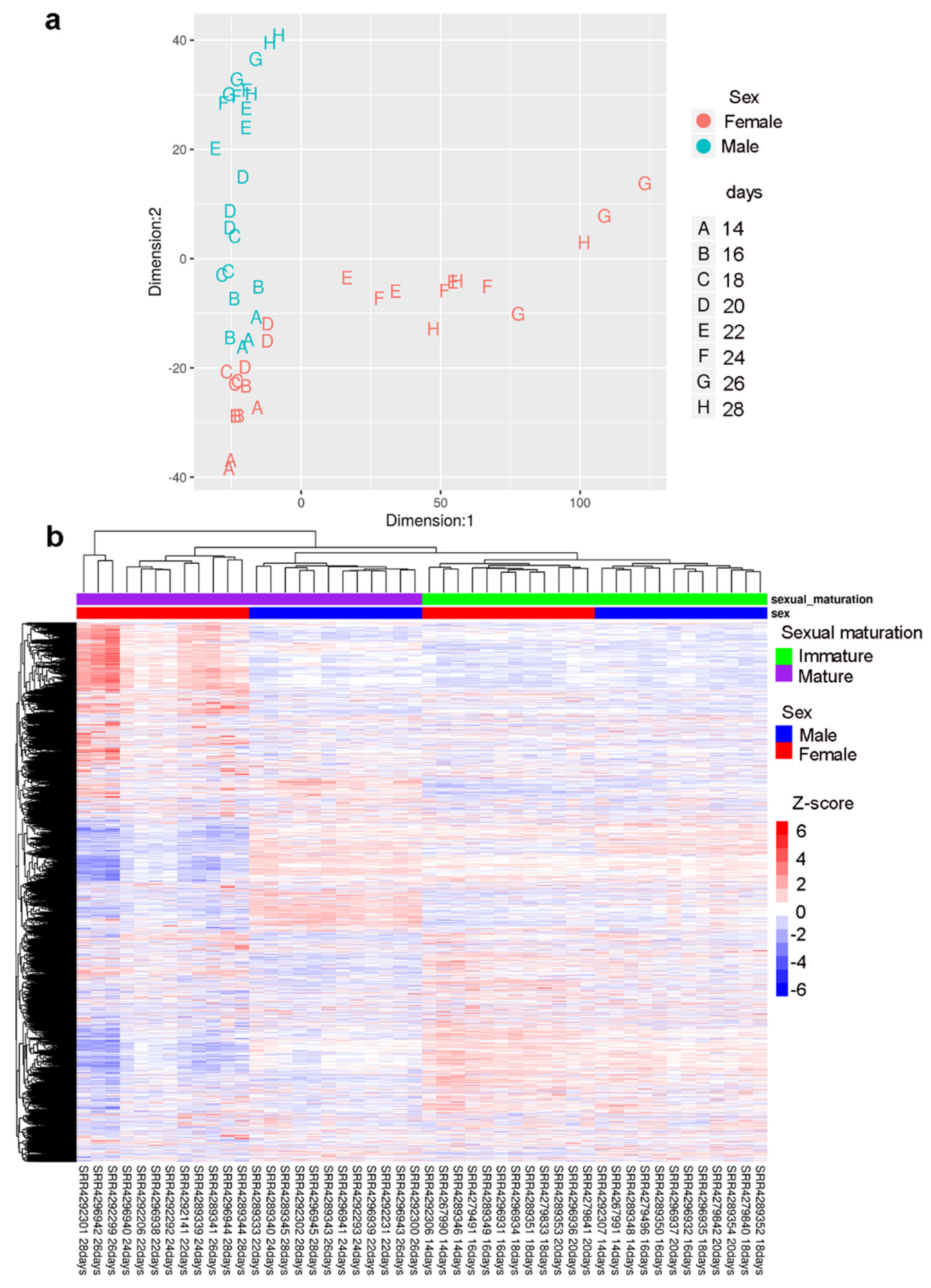
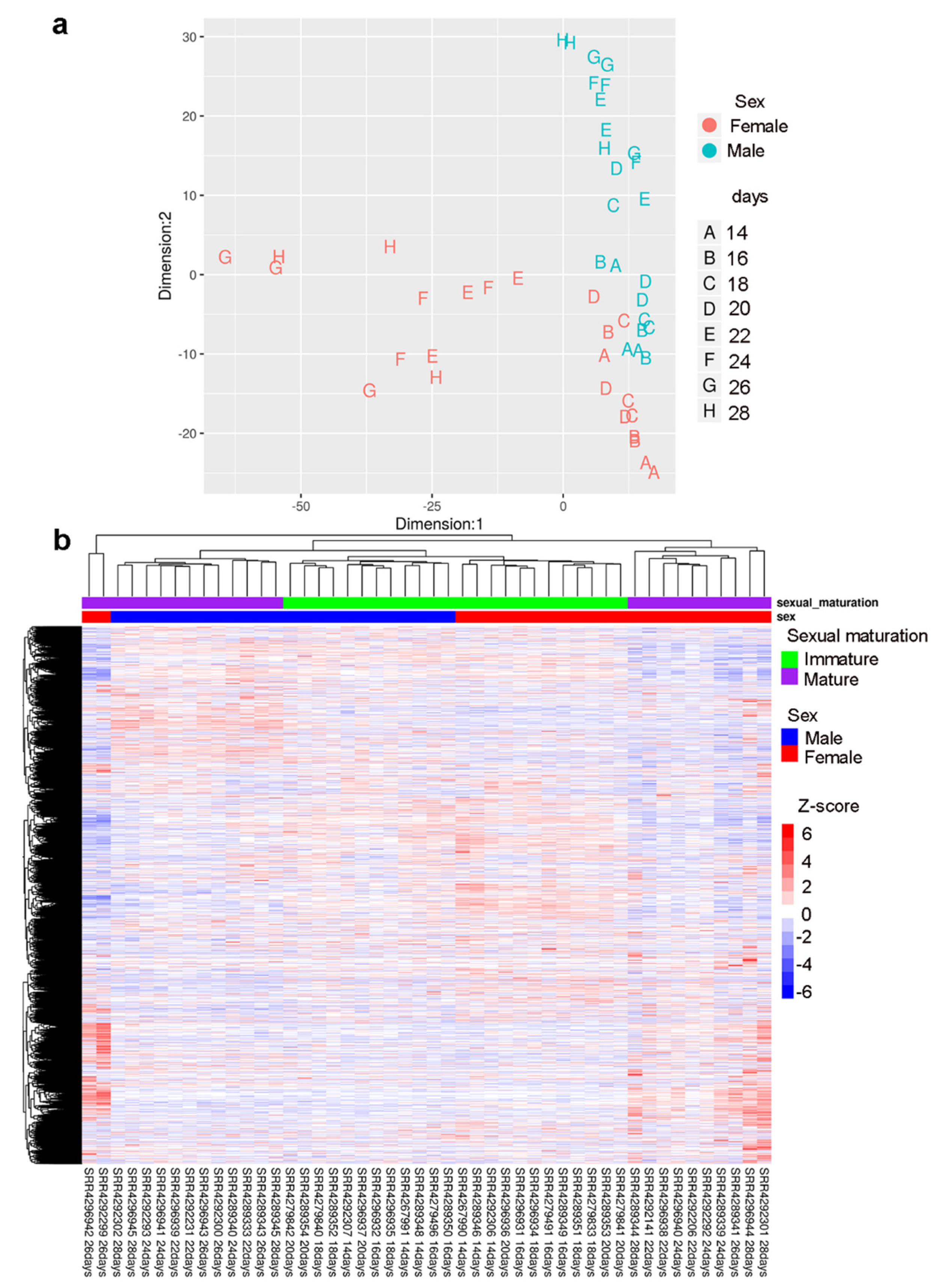
2.3. Gender-Specific lncRNAs Transcriptome
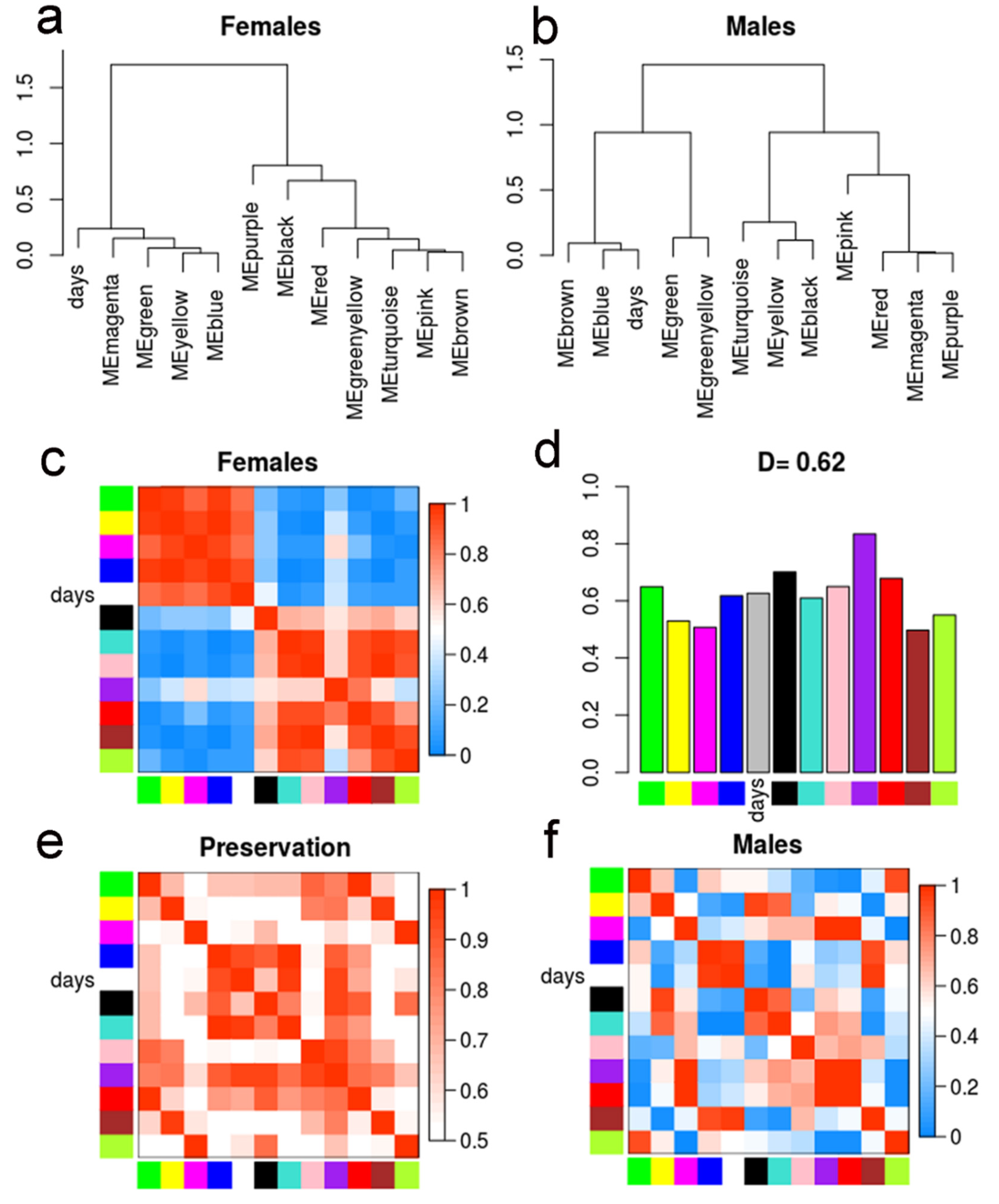
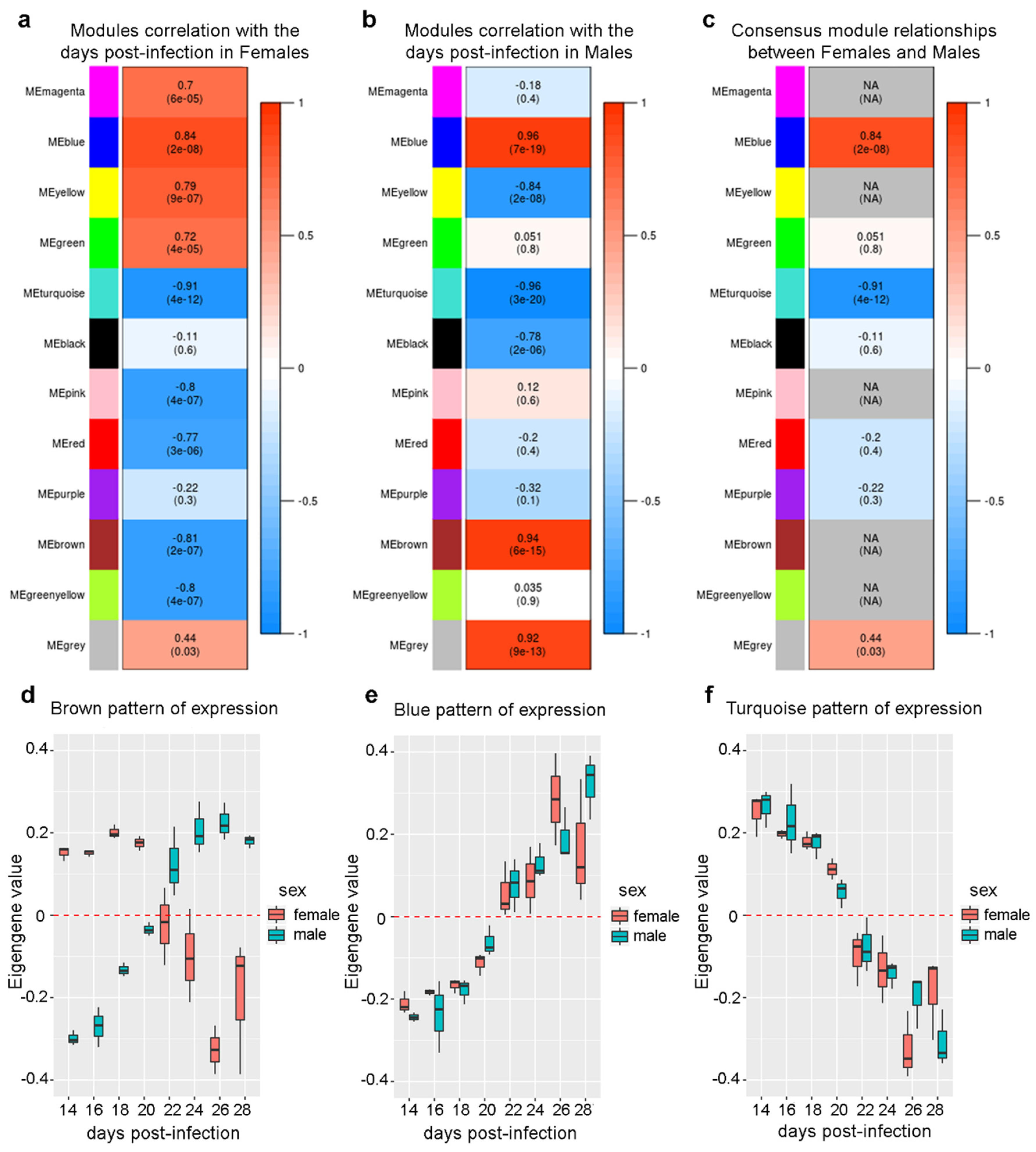
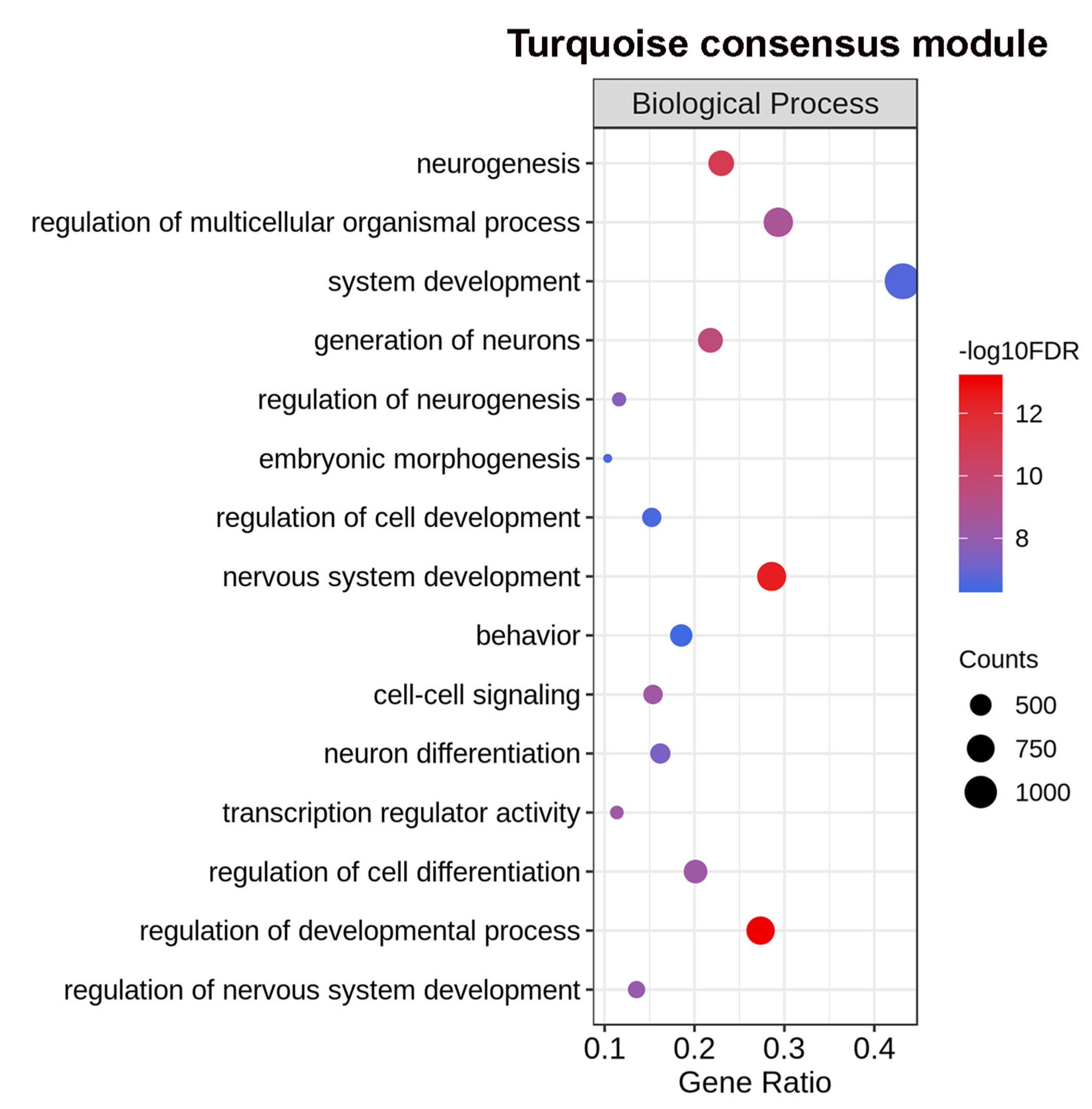
2.4. Differential Eigengene Network Analysis Reveals Different Patterns of Nervous System Differentiation in Males and Females During Sexual Maturation
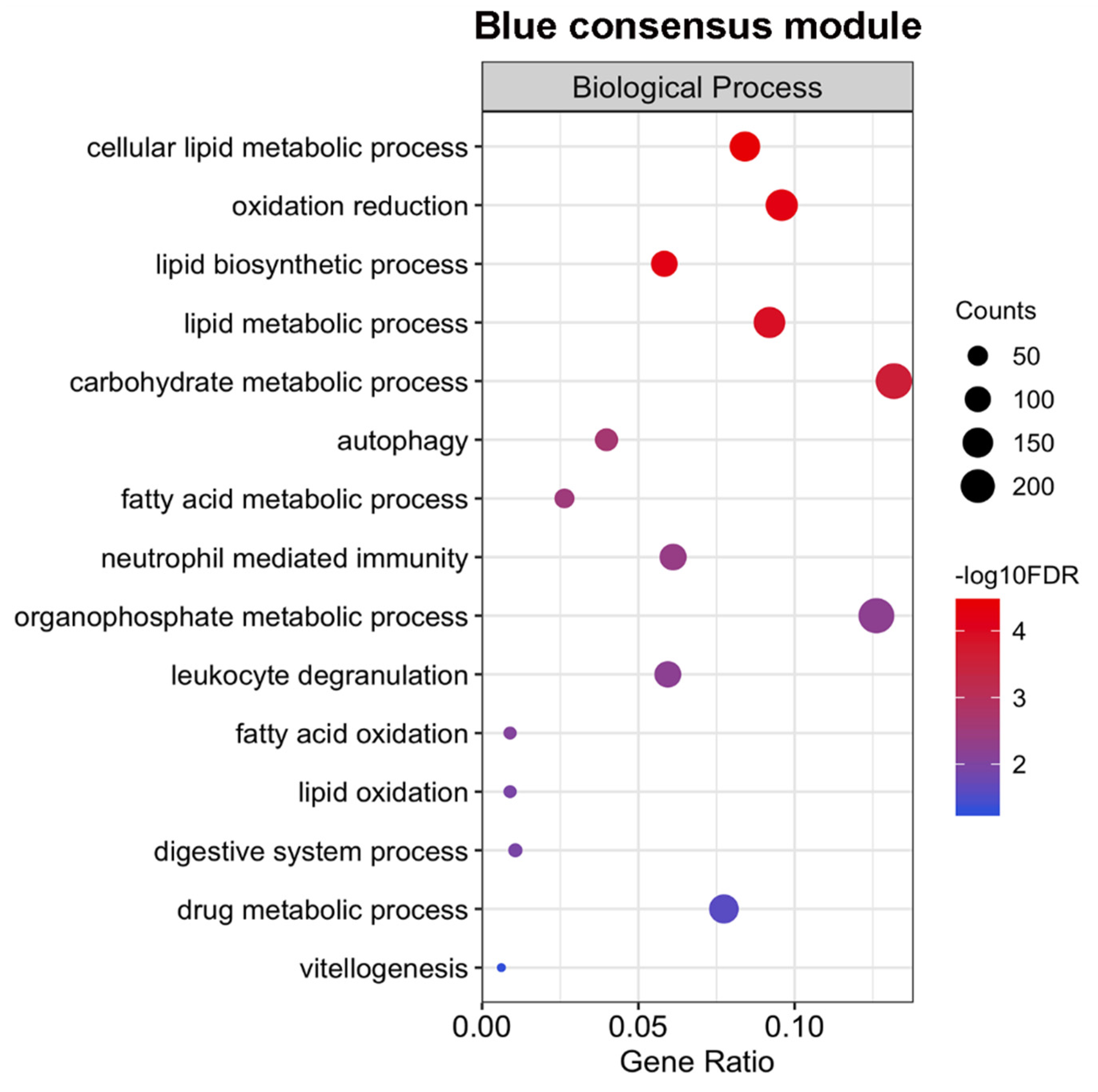
2.5. Expression of Genes Associated with Lipid Metabolism and Host Survival Increases Simultaneously in Males and Females
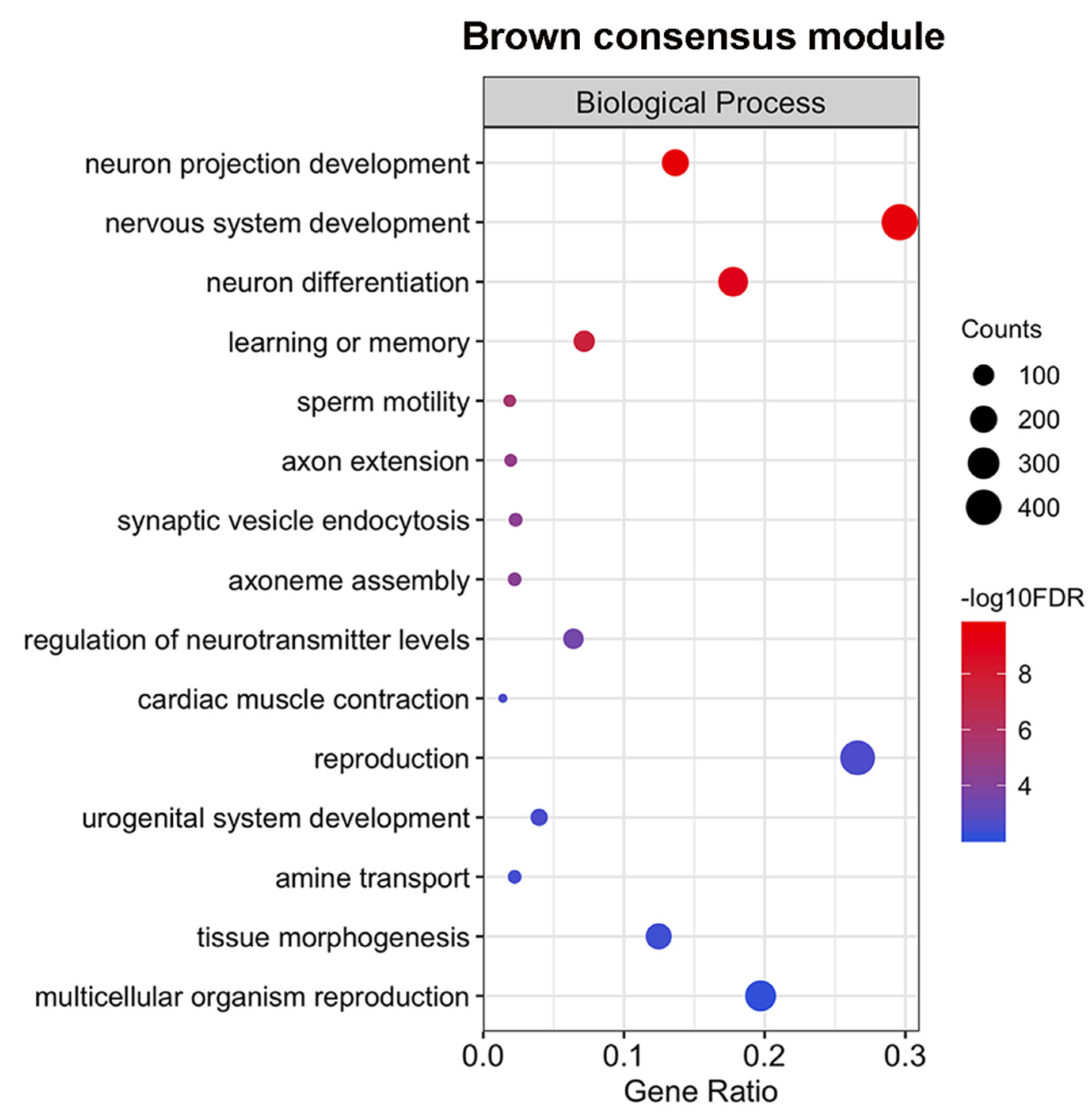
2.6. The Expression of a Large Number of Genes Related to Neurogenesis is Repressed Simultaneously in Males and Females
2.7. Different Sets of Genes Related to Cell Replication and General Nucleotide Metabolism Have Their Expression Changed Only in One Sex Throughout the Sexual Maturation Process
3. Discussion
4. Materials and Methods
4.1. Transcriptome Assembly and lncRNAs Classification
4.2. LncRNAs Conservation Analysis
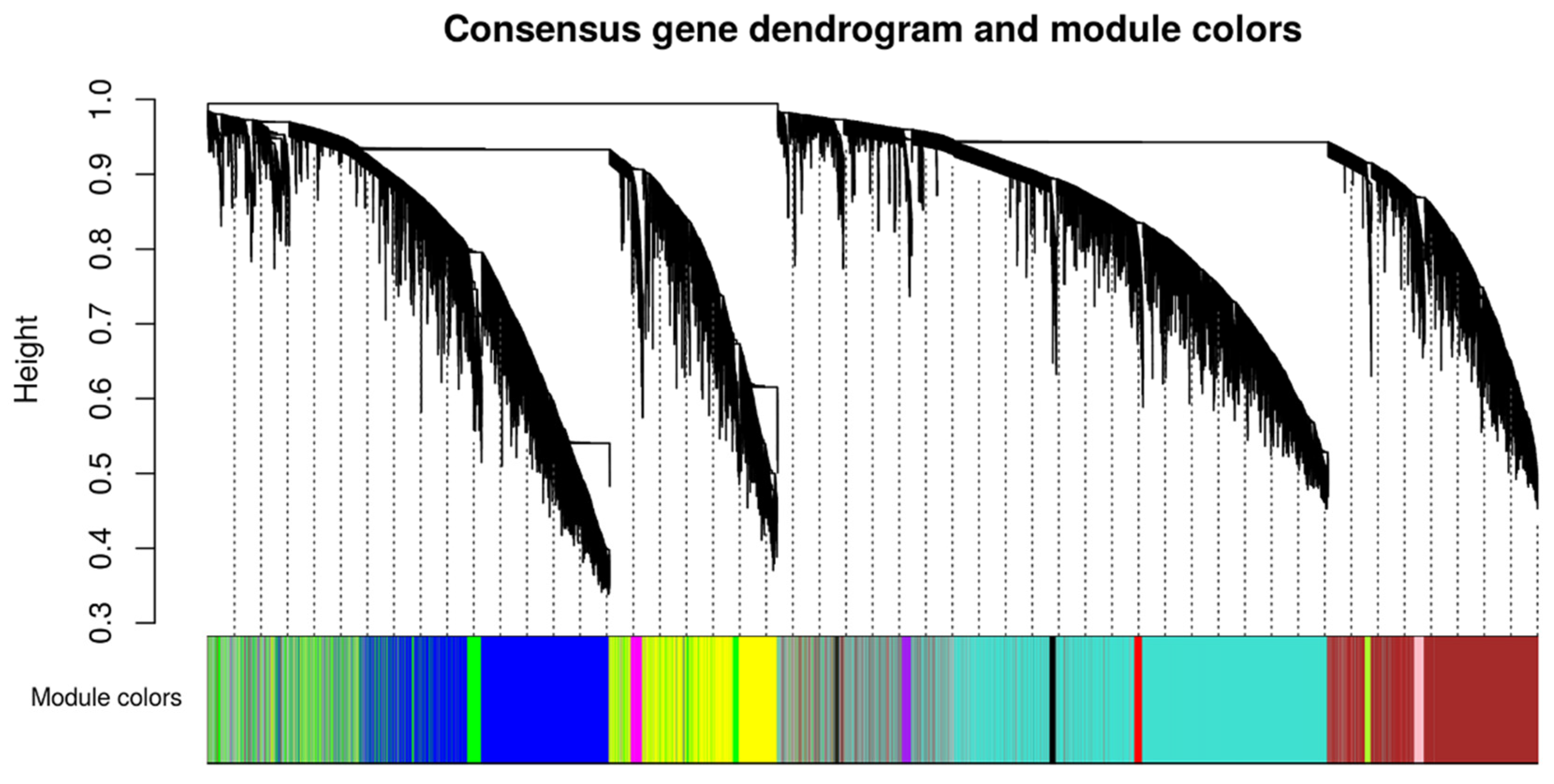
4.3. Weighted Gene Co-Expression Network Analysis
4.4. Gene Ontology (GO) Enrichment
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- World Health Organization. Investing to Overcome the Global Impact of Neglected Tropical Diseases: Third WHO Report on Neglected Tropical Diseases 2015; World Health Organization: Geneva, Switzerland, 2015; Volume 3. [Google Scholar]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Basch, P.F. Intermediate host specificity in Schistosoma mansoni. Exp. Parasitol. 1976, 39, 150–169. [Google Scholar] [CrossRef]
- Lu, Z.; Sessler, F.; Holroyd, N.; Hahnel, S.; Quack, T.; Berriman, M.; Grevelding, C.G. Schistosome sex matters: A deep view into gonad-specific and pairing-dependent transcriptomes reveals a complex gender interplay. Sci. Rep. 2016, 6, 31150. Available online: https://www.nature.com/articles/srep31150#supplementary-information (accessed on 6 January 2020). [CrossRef] [PubMed] [Green Version]
- McManus, D.P.; Dunne, D.W.; Sacko, M.; Utzinger, J.; Vennervald, B.J.; Zhou, X.-N. Schistosomiasis. Nat. Rev. Dis. Primers 2018, 4, 13. [Google Scholar] [CrossRef]
- Cheever, A.W.; Macedonia, J.G.; Mosimann, J.E.; Cheever, E.A. Kinetics of Egg Production and Egg Excretion by Schistosoma mansoni and S. japonicum in Mice Infected with a Single Pair of Worms. Am. J. Trop. Med. Hyg. 1994, 50, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.S.; Mentink-Kane, M.M.; Pesce, J.T.; Ramalingam, T.R.; Thompson, R.; Wynn, T.A. Immunopathology of schistosomiasis. Immunol. Cell Biol. 2007, 85, 148–154. [Google Scholar] [CrossRef]
- Wang, J.; Yu, Y.; Shen, H.; Qing, T.; Zheng, Y.; Li, Q.; Mo, X.; Wang, S.; Li, N.; Chai, R.; et al. Dynamic transcriptomes identify biogenic amines and insect-like hormonal regulation for mediating reproduction in Schistosoma japonicum. Nat. Commun. 2017, 8, 14693. Available online: https://www.nature.com/articles/ncomms14693#supplementary-information (accessed on 6 January 2020). [CrossRef]
- Cao, H.; Wahlestedt, C.; Kapranov, P. Strategies to Annotate and Characterize Long Noncoding RNAs: Advantages and Pitfalls. Trends Genet. 2018, 34, 704–721. [Google Scholar] [CrossRef]
- Liao, Q.; Zhang, Y.; Zhu, Y.; Chen, J.; Dong, C.; Tao, Y.; He, A.; Liu, J.; Wu, Z. Identification of long noncoding RNAs in Schistosoma mansoni and Schistosoma japonicum. Exp. Parasitol. 2018, 191, 82–87. [Google Scholar] [CrossRef]
- Taylor, D.H.; Chu, E.T.-J.; Spektor, R.; Soloway, P.D. Long non-coding RNA regulation of reproduction and development. Mol. Reprod. Dev. 2015, 82, 932–956. [Google Scholar] [CrossRef] [Green Version]
- Maciel, L.F.; Morales-Vicente, D.A.; Silveira, G.O.; Ribeiro, R.O.; Olberg, G.G.O.; Pires, D.S.; Amaral, M.S.; Verjovski-Almeida, S. Weighted Gene Co-Expression Analyses Point to Long Non-Coding RNA Hub Genes at Different Schistosoma mansoni Life-Cycle Stages. Front. Genet. 2019, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Yin, M.; Mo, X.; Sun, C.; Wu, Q.; Zhu, B.; Xiang, M.; Wang, J.; Wang, Y.; Li, J.; et al. An improved genome assembly of the fluke Schistosoma japonicum. PLoS Negl. Trop. Dis. 2019, 13, e0007612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, E.J.R.; daSilva, L.F.; Pires, D.S.; Lavezzo, G.M.; Pereira, A.S.A.; Amaral, M.S.; Verjovski-Almeida, S. The Schistosoma mansoni genome encodes thousands of long non-coding RNAs predicted to be functional at different parasite life-cycle stages. Sci. Rep. 2017, 7, 10508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grutzner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Ubeda, C.; Marin-Barba, M.; Navas-Perez, E.; Gravemeyer, J.; Albuixech-Crespo, B.; Wheeler, G.N.; Garcia-Fernandez, J. Microsyntenic Clusters Reveal Conservation of lncRNAs in Chordates Despite Absence of Sequence Conservation. Biology 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Lefever, S.; Anckaert, J.; Volders, P.-J.; Luypaert, M.; Vandesompele, J.; Mestdagh, P. decodeRNA- predicting non-coding RNA functions using guilt-by-association. Database 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 2007, 1, 54. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Spänig, S.; Weth, O.; Grevelding, C.G. Males, the Wrongly Neglected Partners of the Biologically Unprecedented Male-Female Interaction of Schistosomes. Front. Genet. 2019, 10, 796. [Google Scholar] [CrossRef]
- Pearce, E.J.; Huang, S.C.-C. The metabolic control of schistosome egg production. Cell. Microbiol. 2015, 17, 796–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alencar, A.C.M.D.; Neves, R.H.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Gomes, D.C.; Machado-Silva, J.R. High fat diet has a prominent effect upon the course of chronic schistosomiasis mansoni in mice. Mem. Do Inst. Oswaldo Cruz 2009, 104, 608–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.J., III; King, R.S.; Cogswell, A.; Williams, D.L.; Newmark, P.A. An atlas for Schistosoma mansoni organs and life-cycle stages using cell type-specific markers and confocal microscopy. PLoS Negl. Trop. Dis. 2011, 5, e1009. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Lee, J.; Li, P.; Saberi, A.; Yang, H.; Liu, C.; Zhao, M.; Newmark, P.A. Stem cell heterogeneity drives the parasitic life cycle of Schistosoma mansoni. ELife 2018, 7, e35449. [Google Scholar] [CrossRef] [PubMed]
- Diaz Soria, C.L.; Lee, J.; Chong, T.; Coghlan, A.; Tracey, A.; Young, M.D.; Andrews, T.; Hall, C.; Ng, B.L.; Rawlinson, K.; et al. Single-cell atlas of the first intra-mammalian developmental stage of the human parasite Schistosoma mansoni. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Wangwiwatsin, A.; Protasio, A.V.; Wilson, S.; Owusu, C.; Holroyd, N.E.; Sanders, M.J.; Keane, J.; Doenhoff, M.J.; Rinaldi, G.; Berriman, M. Transcriptome of the parasitic flatworm Schistosoma mansoni during intra-mammalian development. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Pegueroles, C.; Iraola-Guzmán, S.; Chorostecki, U.; Ksiezopolska, E.; Saus, E.; Gabaldón, T. Transcriptomic analyses reveal groups of co-expressed, syntenic lncRNAs in four species of the genus Caenorhabditis. RNA Biol. 2019, 16, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Anguera, M.C.; Ma, W.; Clift, D.; Namekawa, S.; Kelleher, R.J., III; Lee, J.T. Tsx produces a long noncoding RNA and has general functions in the germline, stem cells, and brain. PLoS Genet. 2011, 7, e1002248. [Google Scholar] [CrossRef] [Green Version]
- Golicz, A.A.; Bhalla, P.L.; Singh, M.B. lncRNAs in Plant and Animal Sexual Reproduction. Trends Plant Sci. 2018, 23, 195–205. [Google Scholar] [CrossRef]
- Lawson, H.; Vuong, E.; Miller, R.M.; Kiontke, K.; Fitch, D.H.; Portman, D.S. The Makorin lep-2 and the lncRNA lep-5 regulate lin-28 to schedule sexual maturation of the C. elegans nervous system. ELife 2019, 8, e43660. [Google Scholar] [CrossRef]
- Sarropoulos, I.; Marin, R.; Cardoso-Moreira, M.; Kaessmann, H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature 2019, 571, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokhin, I.; Khorkova, O.; Hsiao, J.; Wahlestedt, C. Developments in lncRNA drug discovery: Where are we heading? Expert Opin. Drug Discov. 2018, 13, 837–849. [Google Scholar] [CrossRef]
- Harries, L.W. RNA Biology Provides New Therapeutic Targets for Human Disease. Front. Genet. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.; Kingsford, C. Accurate assembly of transcripts through phase-preserving graph decomposition. Nat. Biotechnol. 2017, 35, 1167. Available online: https://www.nature.com/articles/nbt.4020#supplementary-information (accessed on 6 January 2020). [CrossRef] [Green Version]
- Niknafs, Y.S.; Pandian, B.; Iyer, H.K.; Chinnaiyan, A.M.; Iyer, M.K. TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat. Methods 2016, 14, 68. Available online: https://www.nature.com/articles/nmeth.4078#supplementary-information (accessed on 6 January 2020). [CrossRef] [Green Version]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Farrer, R.A. Synima: A Synteny imaging tool for annotated genome assemblies. BMC Bioinform. 2017, 18, 507. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Delcher, A.L.; Wortman, J.R.; Salzberg, S.L. DAGchainer: A tool for mining segmental genome duplications and synteny. Bioinformatics 2004, 20, 3643–3646. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module Color | Total Number of Transcripts | Number of lncRNAs | % of lncRNAs |
|---|---|---|---|
| Black | 336 | 82 | 24 |
| Blue | 4465 | 1520 | 34 |
| Brown | 4400 | 1607 | 37 |
| Green | 2288 | 960 | 42 |
| Green-yellow | 128 | 53 | 41 |
| Magenta | 219 | 91 | 42 |
| Pink | 288 | 98 | 34 |
| Purple | 174 | 75 | 43 |
| Red | 521 | 260 | 50 |
| Turquoise | 8161 | 3300 | 40 |
| Yellow | 2830 | 877 | 31 |
| Total | 23,810 | 8923 | 37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciel, L.F.; Morales-Vicente, D.A.; Verjovski-Almeida, S. Dynamic Expression of Long Non-Coding RNAs Throughout Parasite Sexual and Neural Maturation in Schistosoma Japonicum. Non-Coding RNA 2020, 6, 15. https://doi.org/10.3390/ncrna6020015
Maciel LF, Morales-Vicente DA, Verjovski-Almeida S. Dynamic Expression of Long Non-Coding RNAs Throughout Parasite Sexual and Neural Maturation in Schistosoma Japonicum. Non-Coding RNA. 2020; 6(2):15. https://doi.org/10.3390/ncrna6020015
Chicago/Turabian StyleMaciel, Lucas F., David A. Morales-Vicente, and Sergio Verjovski-Almeida. 2020. "Dynamic Expression of Long Non-Coding RNAs Throughout Parasite Sexual and Neural Maturation in Schistosoma Japonicum" Non-Coding RNA 6, no. 2: 15. https://doi.org/10.3390/ncrna6020015
APA StyleMaciel, L. F., Morales-Vicente, D. A., & Verjovski-Almeida, S. (2020). Dynamic Expression of Long Non-Coding RNAs Throughout Parasite Sexual and Neural Maturation in Schistosoma Japonicum. Non-Coding RNA, 6(2), 15. https://doi.org/10.3390/ncrna6020015