
Zooming in: PAGE-Northern Blot Helps to Analyze Anti-Sense Transcripts Originating from Human rIGS under Transcriptional Stress


Abstract
:1. Introduction
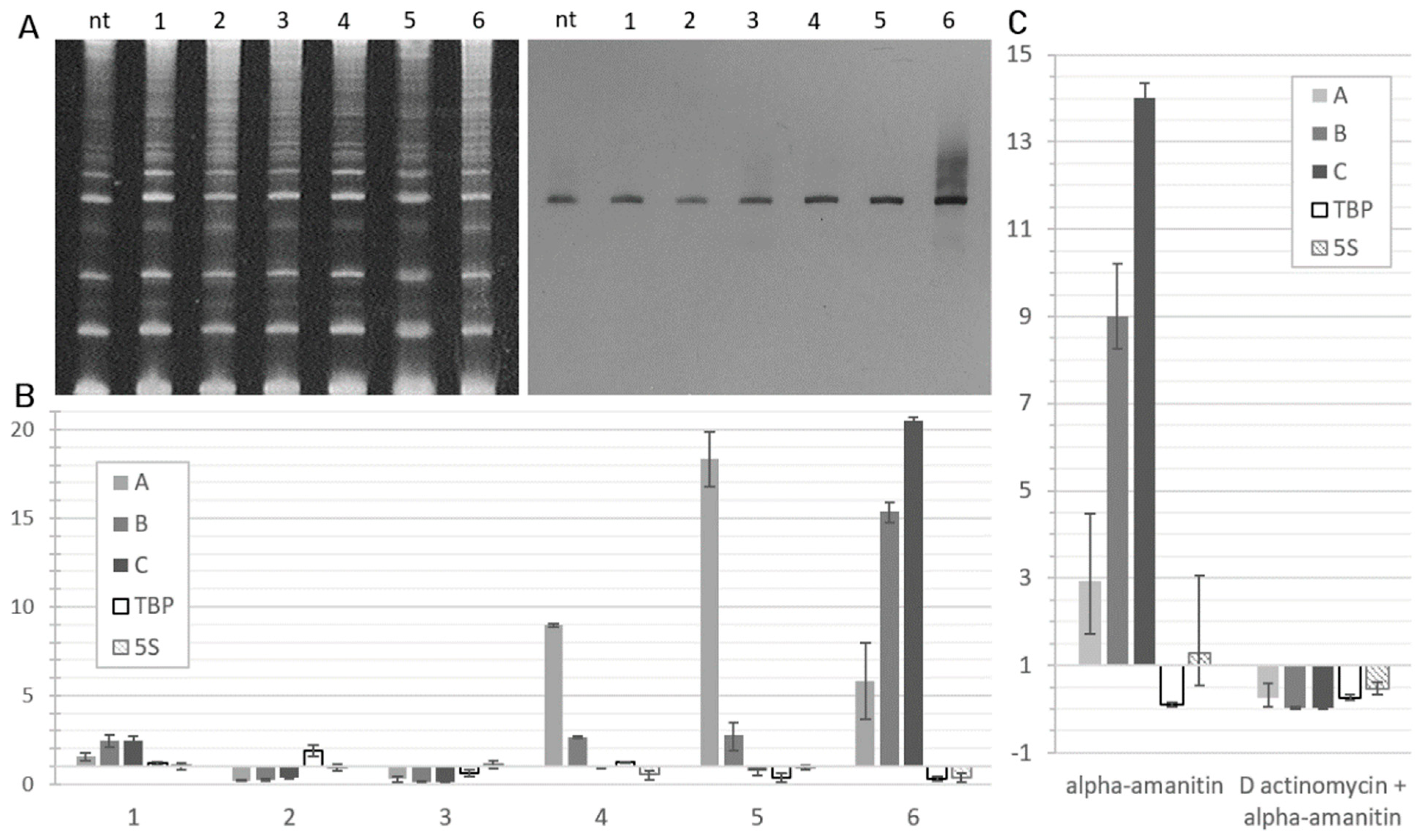
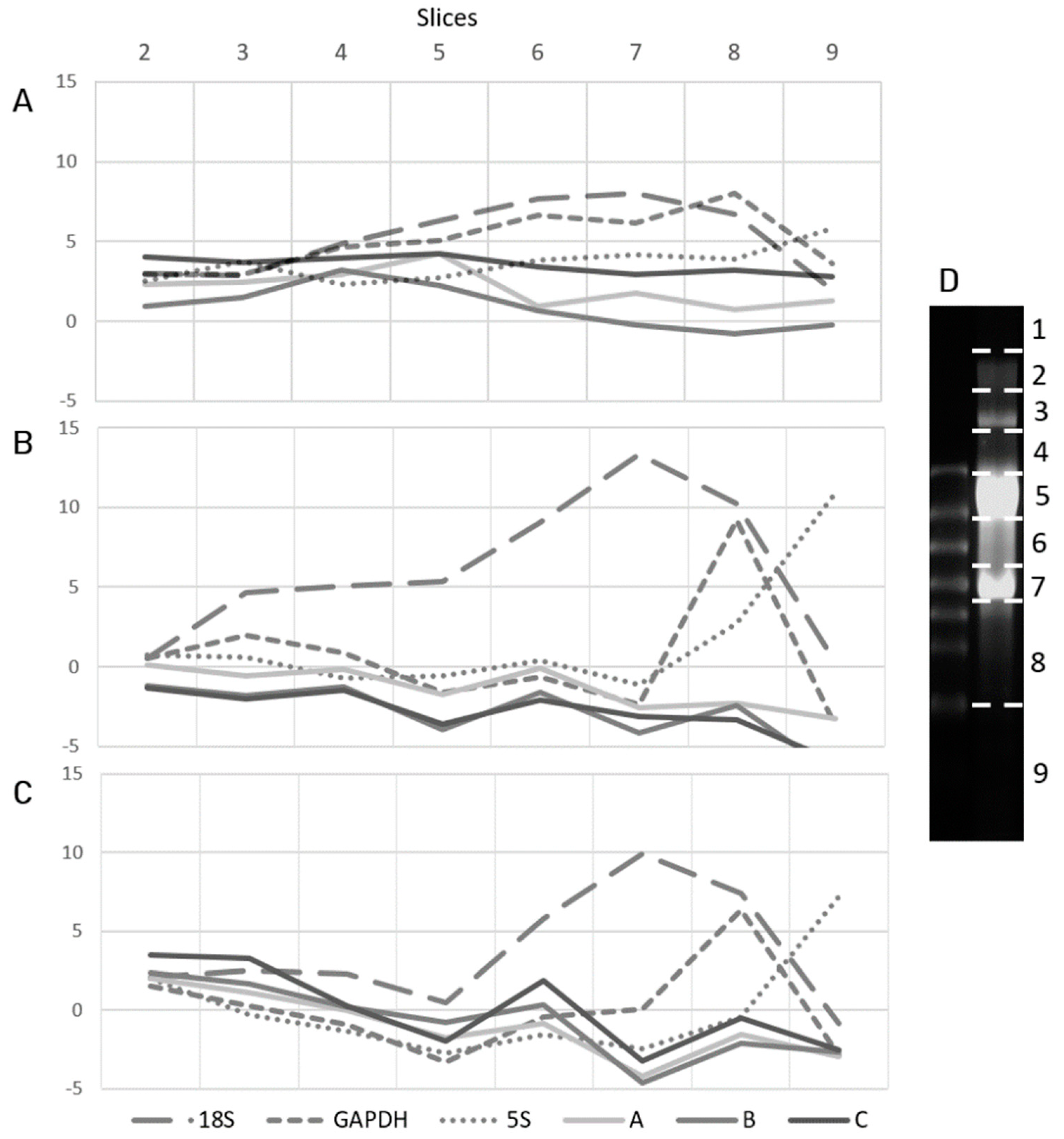
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Alpha-Amanitin Treatment
4.2. RNA Extraction from Cells
4.3. Northern Blotting
4.4. Reverse Transcription and Quantitative PCR
4.5. rRNA Depletion and Slices Preparation
4.6. Statistics
4.7. PAGE Northern Blot
4.8. 3′RACE and Southern Blot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence, 5′-3′ | Amplicon Size, bp |
|---|---|---|
| A | GTTTCGTCCTTTTGAGACAGAGT, f GTGGGCGCATCACAGGAGGTC, r | 254 |
| B | TCCAACTCCCGACCTCCTGT, f TGCAGAGATACACGTTGTCGTTG, r | 194 |
| C | CAACGACAACGTGTATCTCTGCA, f TACGCTCGGTTCATTTACACACA, r | 209 |
| 18S | TTTCTCGATTCCGTGGGTGG, f CCCGGACATCTAAGGGCATC, r | 211 |
| 5S (from [49], modified) | GGCCATACCACCCTGAACG, f CAGCACCCGGTATTCCCAG, r | 107 |
| TBP | TCCCATGACCCCCATCACTC, f ATATTCGGCGTTTCGGGCA, r | 145 |
| GAPDH | ACCGTCAAGGCTGAGAACG, f GCATCGCCCCACTTGATTTT, r | 95 |
| 3′RACE adapter | GCGAGCACAGAATTAATACGACTCACTATAGGT12VN | - |
| 3′RACE primer | GCGAGCACAGAATTAATACGACT | |
| ABC sense RNA probe | GUUUCGUCCUUUUGAGACAGAGUUUCACUCUUGUUUCCACGGCUAGAGU GCAAUGGCGCGAUCUUGGCUCACCGCACCUUCCGCCUCCCGGGUUCGAGCGCUUCUCCUGCCUCCAGCCUCCCGAUUAGCGGGGAUUGACAGGGAGGCACCCCCACGCCUGGCUUGGCUGAUGUUUGUGUUUUUAGUAGGCACGCCGUGUCUCUCCAUGUUGCUCAGGCUGGUCUCCAACUCCCGACCUCCUGUGAUGCGCCCACCUCGGCCUCUCGAAGUGCUGGGAUGACGGGCGUGACGACCGUGCCCGGCCUGUUGACUCAUUUCGCUUUUUUAUUUCUUUCGUUUCCACGCGUUUACUUAUAUGUAUUAAUGUAAACGUUUCUGUACGCUUAUAUGCAAACAACGACAACGUGUAUCUCUGCAUUGAAUACUCUUGCGUAUGGUAAAUACGUAUCGGUUGUAUGGAAAUAGACUUCUGUAUGAUAGAUGUAGGUGUCUGUGUUAUACAAAUAAAUACACAUCGCUCUAUAAAGAAGGGAUCGUCGAUAAAGACGUUUAUUUUACGUAUGAAAAGCGUCGUAUUUAUGUGUGUAAAUGAACCGAGCGUA | |
| ABC anti-sense RNA probe | UACGCUCGGUUCAUUUACACACAUAAAUACGACGCUUUUCAUACGUAAAAUAAACGUCUUUAUCGACGAUCCCUUCUUUAUAGAGCGAUGUGUAUUUAUUUGUAUAACACAGACACCUACAUCUAUCAUACAGAAGUCUAUUUCCAUACAACCGAUACGUAUUUACCAUACGCAAGAGUAUUCAAUGCAGAGAUACACGUUGUCGUUGUUUGCAUAUAAGCGUACAGAAACGUUUACAUUAAUACAUAUAAGUAAACGCGUGGAAACGAAAGAAAUAAAAAAGCGAAAUGAGUCAACAGGCCGGGCACGGUCGUCACGCCCGUCAUCCCAGCACUUCGAGAGGCCGAGGUGGGCGCAUCACAGGAGGUCGGGAGUUGGAGACCAGCCUGAGCAACAUGGAGAGACACGGCGUGCCUACUAAAAACACAAACAUCAGCCAAGCCAGGCGUGGGGGUGCCUCCCUGUCAAUCCCCGCUAAUCGGGAGGCUGGAGGCAGGAGAAGCGCUCGAACCCGGGAGGCGGAAGGUGCGGUGAGCCAAGAUCGCGCCAUUGCACUCUAGCCGUGGAAACAAGAGUGAAACUCUGUCUCAAAAGGACGAAAC | |
| ABC DNA probe | GTTTCGTCCTTTTGAGACAGAGTTTCACTCTTGTTTCCACGGCTAGAGTGCAATGGCGCGATCTTGGCTCACCGCACCTTCCGCCTCCCGGGTTCGAGCGCTTCTCCTGCCTCCAGCCTCCCGATTAGCGGGGATTGACAGGGAGGCACCCCCACGCCTGGCTTGGCTGATGTTTGTGTTTTTAGTAGGCACGCCGTGTCTCTCCATGTTGCTCAGGCTGGTCTCCAACTCCCGACCTCCTGTGATGCGCCCACCTCGGCCTCTCGAAGTGCTGGGATGACGGGCGTGACGACCGTGCCCGGCCTGTTGACTCATTTCGCTTTTTTATTTCTTTCGTTTCCACGCGTTTACTTATATGTATTAATGTAAACGTTTCTGTACGCTTATATGCAAACAACGACAACGTGTATCTCTGCATTGAATACTCTTGCGTATGGTAAATACGTATCGGTTGTATGGAAATAGACTTCTGTATGATAGATGTAGGTGTCTGTGTTATACAAATAAATACACATCGCTCTATAAAGAAGGGATCGTCGATAAAGACGTTTATTTTACGTATGAAAAGCGTCGTATTTATGTGTGTAAATGAACCGAGCGTA |
References
- Kim, V.N. Small RNAs: Classification, biogenesis, and function. Mol. Cells 2005, 19, 1–15. [Google Scholar] [PubMed]
- Thairu, M.W.; Hansen, A.K. It’s a small, small world: Unravelling the role and evolution of small RNAs in organelle and endosymbiont genomes. FEMS Microbiol. Lett. 2019, 366, fnz049. [Google Scholar] [CrossRef]
- Chen, G.; Shi, T.; Shi, L. Characterizing and annotating the genome using RNA-seq data. Sci. China Life Sci. 2016, 60, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Backofen, R.; Engelhardt, J.; Erxleben, A.; Fallmann, J.; Grüning, B.; Ohler, U.; Rajewsky, N.; Stadler, P.F. RNA-bioinformatics: Tools, services and databases for the analysis of RNA-based regulation. J. Biotechnol. 2017, 261, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P.; Christopoulos, T.K. The biotin-(strept)avidin system: Principles and applications in biotechnology. Clin. Chem. 1991, 37, 625–636. [Google Scholar] [CrossRef]
- Rosemeyer, V.; Laubrock, A.; Seibl, R. Nonradioactive 3′-End-Labeling of RNA Molecules of Different Lengths by Terminal Deoxynucleotidyltransferase. Anal. Biochem. 1995, 224, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.P.G.; Qin, P.Z. A facile method for attaching nitroxide spin labels at the 5′ terminus of nucleic acids. Nucleic Acids Res. 2007, 35, e77. [Google Scholar] [CrossRef] [Green Version]
- Tijssen, P. (Ed.) Chapter 7—Labeling of probes and their detection. In Laboratory Techniques in Biochemistry and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 1993; Volume 24, pp. 269–374. [Google Scholar]
- Nilsen, T.W. Gel Purification of RNA. Cold Spring Harb. Protoc. 2013, 2013, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papakonstantinou, T.; Zhang, C. Purification of Nucleic Acids from Gels. In Handbook of Nucleic Acid Purification; CRC Press: Boca Raton, FL, USA, 2009; pp. 537–555. [Google Scholar]
- Petrov, A.; Wu, T.; Puglisi, E.V.; Puglisi, J.D. RNA Purification by Preparative Polyacrylamide Gel Electrophoresis. Methods Enzymol. 2013, 530, 315–330. [Google Scholar] [CrossRef]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersaglieri, C.; Santoro, R. Genome Organization in and around the Nucleolus. Cells 2019, 8, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eickbush, T.H.; Eickbush, D.G. Finely Orchestrated Movements: Evolution of the Ribosomal RNA Genes. Genetics 2007, 175, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Agrawa, S.; Ganley, A.R.D. The conservation landscape of the human ribosomal RNA gene repeats. PLoS ONE 2018, 13, e0207531. [Google Scholar]
- Audas, T.; Jacob, M.D.; Lee, S. Immobilization of Proteins in the Nucleolus by Ribosomal Intergenic Spacer Noncoding RNA. Mol. Cell 2012, 45, 147–157. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.-Y.; Wan, F.-C.; Liu, F.-J.; Liu, J.; Zhang, N.; Jin, S.-H.; Li, J.-Y. Deep sequencing analysis of small non-coding RNAs reveals the diversity of microRNAs and piRNAs in the human epididymis. Gene 2012, 497, 330–335. [Google Scholar] [CrossRef]
- Mayer, C.; Schmitz, K.-M.; Li, J.; Grummt, I.; Santoro, R. Intergenic Transcripts Regulate the Epigenetic State of rRNA Genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Mars, J.C.; Sabourin-Felix, M.; Tremblay, M.G.; Moss, T. Deconvolution Protocol for ChIP-Seq Reveals Analogous Enhancer Structures on the Mouse and Human Ribosomal RNA Genes. G3 Genes Genomes Genet. 2018, 8, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Shiao, Y.-H.; Lupascu, S.T.; Gu, Y.D.; Kasprzak, W.; Hwang, C.J.; Fields, J.R.; Leighty, R.M.; Quiñones, O.; Shapiro, B.A.; Alvord, W.G.; et al. An Intergenic Non-Coding rRNA Correlated with Expression of the rRNA and Frequency of an rRNA Single Nucleotide Polymorphism in Lung Cancer Cells. PLoS ONE 2009, 4, e7505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadova, A.A.; Kupriyanova, N.S.; Pavlova, G.V. Mapping and Quantification of Non-Coding RNA Originating from the rDNA in Human Glioma Cells. Cancers 2020, 12, 2090. [Google Scholar] [CrossRef]
- Zentner, G.; Saiakhova, A.; Manaenkov, P.; Adams, M.D.; Scacheri, P.C. Integrative genomic analysis of human ribosomal DNA. Nucleic Acids Res. 2011, 39, 4949–4960. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mueller, J.E.; Bryk, M. Sir2 represses endogenous polymerase II transcription units in the ribosomal DNA non-transcribed spacer. Mol. Biol. Cell 2006, 17, 3848–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, K.J.; Khosraviani, N.; Chan, J.N.Y.; Gorthi, A.; Samman, A.; Zhao, D.Y.; Wang, M.; Bokros, M.; Vidya, E.; Ostrowski, L.A.; et al. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 2020, 585, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Khitrinskaia, I.; Stepanov, V.A.; Puzyrev, V.P. Alu repeats in the human genome. Mol. Biol. 2003, 3, 382–391. [Google Scholar]
- Zhang, X.-O.; Gingeras, T.R.; Weng, Z. Genome-wide analysis of polymerase III–transcribed Alu elements suggests cell-type–specific enhancer function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Pan, J.; Thoroddsen, V.; Wysong, D.R.; Blackman, R.K.; Bulawa, C.E.; Gould, A.E.; Ocain, T.D.; Dick, L.R.; Errada, P.; et al. Novel Small-Molecule Inhibitors of RNA Polymerase III. Eukaryot. Cell 2003, 2, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules 2015, 20, 3898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Miranda-Gonçalves, V.; Camilo, V.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; et al. Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors. Cancers 2020, 12, 2903. [Google Scholar] [CrossRef]
- Sobell, H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. USA 1985, 82, 5328–5331. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.L.; Veiga, S.R.; Almacellas, E.; Hernandez-Losa, J.; Ferreres, J.C.; Kozma, S.C.; Ambrosio, S.; Thomas, G.V.; Tauler, A. Effect of low doses of actinomycin D on neuroblastoma cell lines. Mol. Cancer 2016, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2012, 65, 157–170. [Google Scholar] [CrossRef]
- Colis, L.; Peltonen, K.D.; Sirajuddin, P.; Liu, H.; Sanders, S.; Ernst, G.; Barrow, J.; Laiho, M. DNA intercalator BMH-21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget 2014, 5, 4361–4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, A.V.; Lemos, B. Ribosomal DNA and the Nucleolus as Keystones of Nuclear Architecture, Organization, and Function. Trends Genet. 2019, 35, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Fedoseeva, D.M.; Klushevskaya, E.S.; Slovohotov, I.Y.; Chechetkin, V.R.; Kravatsky, Y.V.; Kretova, O.V. rDNA Clusters Make Contact with Genes that Are Involved in Differentiation and Cancer and Change Contacts after Heat Shock Treatment. Cells 2019, 8, 1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, D.J.; Huang, S. Regulation of ribosome biogenesis within the nucleolus. FEBS Lett. 2001, 509, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Matos-Perdomo, E.; Machín, F. Nucleolar and Ribosomal DNA Structure under Stress: Yeast Lessons for Aging and Cancer. Cells 2019, 8, 779. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, M.; Fujii, Y.R. Human Ribosomal RNA-Derived Resident MicroRNAs as the Transmitter of Information upon the Cytoplasmic Cancer Stress. BioMed Res. Int. 2016, 2016, 7562085. [Google Scholar] [CrossRef] [Green Version]
- Duttke, S.H. RNA Polymerase III Accurately Initiates Transcription from RNA Polymerase II Promoters in Vitro. J. Biol. Chem. 2014, 289, 20396–20404. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. RNA Polymerase II Activity of Type 3 Pol III Promoters. Mol. Ther. Nucleic Acids 2018, 12, 135–145. [Google Scholar] [CrossRef]
- Ullu, E.; Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 1984, 312, 171–172. [Google Scholar] [CrossRef]
- Dieci, G.; Conti, A.; Pagano, A.; Carnevali, D. Identification of RNA polymerase III-transcribed genes in eukaryotic genomes. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2013, 1829, 296–305. [Google Scholar] [CrossRef]
- Kravchenko, J.E.; Rogozin, I.; Koonin, E.V.; Chumakov, P. Transcription of mammalian messenger RNAs by a nuclear RNA polymerase of mitochondrial origin. Nature 2005, 436, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.L.; Chiao, C.H.; Hsu, M.T. Transcription of muscle actin genes by a nuclear form of mitochondrial RNA polymerase. PLoS ONE 2011, 6, e22583. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2015, 1859, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzi, L.; Chiu, H.-S.; Cobos, F.A.; Gross, S.; Volders, P.-J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 2021, 1–13. [Google Scholar] [CrossRef]
- Aranda, P.S.; Lajoie, D.M.; Jorcyk, C.L. Bleach gel: A simple agarose gel for analyzing RNA quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef] [Green Version]
- Shor, B.; Wu, J.; Shakey, Q.; Toral-Barza, L.; Shi, C.; Follettie, M.; Yu, K. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA poly-merase III-dependent transcription in cancer cells. J. Biol. Chem. 2010, 285, 15380–15392. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadova, A.A.; Panteleev, D.Y.; Pavlova, G.V. Zooming in: PAGE-Northern Blot Helps to Analyze Anti-Sense Transcripts Originating from Human rIGS under Transcriptional Stress. Non-Coding RNA 2021, 7, 50. https://doi.org/10.3390/ncrna7030050
Sadova AA, Panteleev DY, Pavlova GV. Zooming in: PAGE-Northern Blot Helps to Analyze Anti-Sense Transcripts Originating from Human rIGS under Transcriptional Stress. Non-Coding RNA. 2021; 7(3):50. https://doi.org/10.3390/ncrna7030050
Chicago/Turabian StyleSadova, Anastasia A., Dmitry Y. Panteleev, and Galina V. Pavlova. 2021. "Zooming in: PAGE-Northern Blot Helps to Analyze Anti-Sense Transcripts Originating from Human rIGS under Transcriptional Stress" Non-Coding RNA 7, no. 3: 50. https://doi.org/10.3390/ncrna7030050
APA StyleSadova, A. A., Panteleev, D. Y., & Pavlova, G. V. (2021). Zooming in: PAGE-Northern Blot Helps to Analyze Anti-Sense Transcripts Originating from Human rIGS under Transcriptional Stress. Non-Coding RNA, 7(3), 50. https://doi.org/10.3390/ncrna7030050