
Common Features in lncRNA Annotation and Classification: A Survey

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Commonly Used Features in Non-Coding Transcript Annotation
- k-mers are k-letter subsequences of a given sequence stretch. A single base is a 1-mer, whereas codons are base-triplets and thus 3-mers. The information encoded by k-mers is distributed based on the value of k. Shorter k-mers are more abundant and their relative frequencies are more strongly cross-correlated than for longer k-mers. A 7-mer would encode more information than a 3-mer, for example, as the probability of occurrence of a particular 7-mer is much lower than that of a 3-mer. However, frequencies of longer k-mers become computationally more expensive to calculate [18,19]: For there are already distinct features, which translates to an at least equal number of subsequent comparison operations. In addition to the computational issues, sufficiently large training sets and test sets are required.
- Euclidean and logarithmic distances of the frequency vectors of certain features such as k-mer frequencies relative to the expected values of these frequencies in a reference set of lncRNAs and protein-coding sequences, respectively, are utilized in, e.g., LncFinder [20].
- GC content is the number of purine bases (either G or C) in the sequence normalized by the length of the sequence. Higher GC content tends to be closely associated with the presence of RNA secondary structure which in turn tends to correlate with biological function aside from the encoding of proteins [21]. The GC content can therefore serve as an indicator for coding potential.
- Fickett TESTCODE was the first method proposed to find a distinguishing factor between the two classes of RNA [22]. To circumvent the hard problem of identifying initiation signals in a sequence, the authors devised a test to identify whether a given piece of DNA or RNA is coding or non-coding directly from the sequence. It is based on the asymmetric distribution of bases in protein-coding sequences. The test is based on eight different parameters, the first four being measures of the bases A, T, G and C, calculating the probabilities for them being favored in one of the three codon positions. The latter four are the ratios of each base in the sequence. Attaching weights to each parameter, the coding potential is computed, called TESTCODE or Fickett score.The detection tool CPAT [23], derives the probability of a base being favored at a certain position as:The derived values are then converted into probabilities (p) using the lookup table provided by Fickett [22] or an updated table taking into account newly annotated transcripts, as provided for example by Wang et al. [23]. In principle, this gives a measure for how non-random a nucleotide is distributed across 3-mers of a given sequence. Furthermore, w in Equation (1) is a weight representing the predictive power of each parameter on its own given sequences with an already known function. It has been shown that the Ficket score can achieve 94% and 97% sensitivity and specificity, respectively, on lncRNA sequences, being inconclusive for 18% of the sequences [23].
- Hexamer score. Adjacent amino acids in a protein are not independent [23], especially with regards to their direct neighbors. This results in a hexamer usage bias that can be used to calculate a relative score. Coding sequences usually derive a positive, non-coding sequences a negative score [24]. There are several ways of defining a hexamer score. The strategy used in CPAT [23] computes the log-likelihood ratio between coding and non-coding sequences. The score for a sequence S = with m hexamers is derived as:where and represent the probability of each hexamer to be part of a protein-coding and non-coding sequence, respectively, with 4096 total hexamers possible.
- ORF length. Translation of mRNAs into proteins by the ribosome begins at a start codon (AUG) and ends at one of the stop codons (UAA/UGA/UAG) of spliced mRNA. Thus, ORFs are the theoretically translatable subunits of genes and therefore of functional relevance. The length of a putative ORF is a feature frequently used to predict if a sequence has any coding potential [12]. It should be noted that not every ORF translatable on a sequence level is in fact translated to a peptide chain.
- ORF coverage. The ratio between the ORF length and the length of the sequence in consideration is used. Long ORFs are generally considered as an indicator of a coding sequence. Following that logic, a low coverage indicates a non-coding sequence [25].
- Peptide length. The hypothetically translated amino acid (AA) sequence of a given ORF can be analyzed for physico-chemical and general properties. The length of this AA-sequence corresponds to the peptide length. Note that this property is obviously linked to the ORF length.
- Hydropathy is a measure of hydrophilic or hydrophobic interactions of a potential peptide sequence with a water-based solvent. Since amino acids are categorized as either hydrophobic, hydrophilic or neutral, this property also implicitly contains information regarding the AA composition of a protein. A hydropathic index is typically calculated as a contribution function from all individual amino acid residues present in a peptide chain, see [26] for a review of amino acid hydrophobicity scales.
- Isoelectric point: The isoelectric point (pI) refers to the pH value where a balance between negative and positive charges in an amphoteric molecule is reached. As practically all relevant proteins are amphoteric, the pI is a relevant measure for the AA composition. Different residues contribute different physico-chemical properties depending on their free electron pairs. As with the hydropathic score the pI is usually approximated using the contribution of all present residues in a given peptide sequence, see, e.g., [27].
- PolyA abundance: The presence of polyadenylation signals in a transcript is a strong indicator for a protein coding function. Polyadenylation typically happens at the 3′-terminus of mature mRNAs in eukaryotic cells. It is suggested to increase stability of the transcript while simultaneously regulating its durability. The proportion of PolyA signals (5′-AAUAAA-3′) as a fraction of sequence length can be used a measure of coding potential.
- RNA minimum free energy (MFE) is often used as a metric for the inherent stability of the RNA secondary structure of a given nucleotide sequence and refers to the thermodynamic energy of the most stable conformation. Although the MFE does not necessarily correspond to the true RNA secondary structure adopted in vivo it is often used as reasonable approximation. Since non-coding RNAs tend to have a much higher degree of conserved structural complexity, the MFE can be used to infer sequences with a comparably low coding potential.
3. Contemporary lncRNA Detection Strategies
3.1. Strategies Based on Subsequence Decomposition
3.2. Approaches Considering Open Reading Frames (ORF) and Most-Likely Coding Sequences
3.3. Approaches Using Potential Protein Features
3.4. Tools Utilizing RNA Secondary Structure Properties as Features
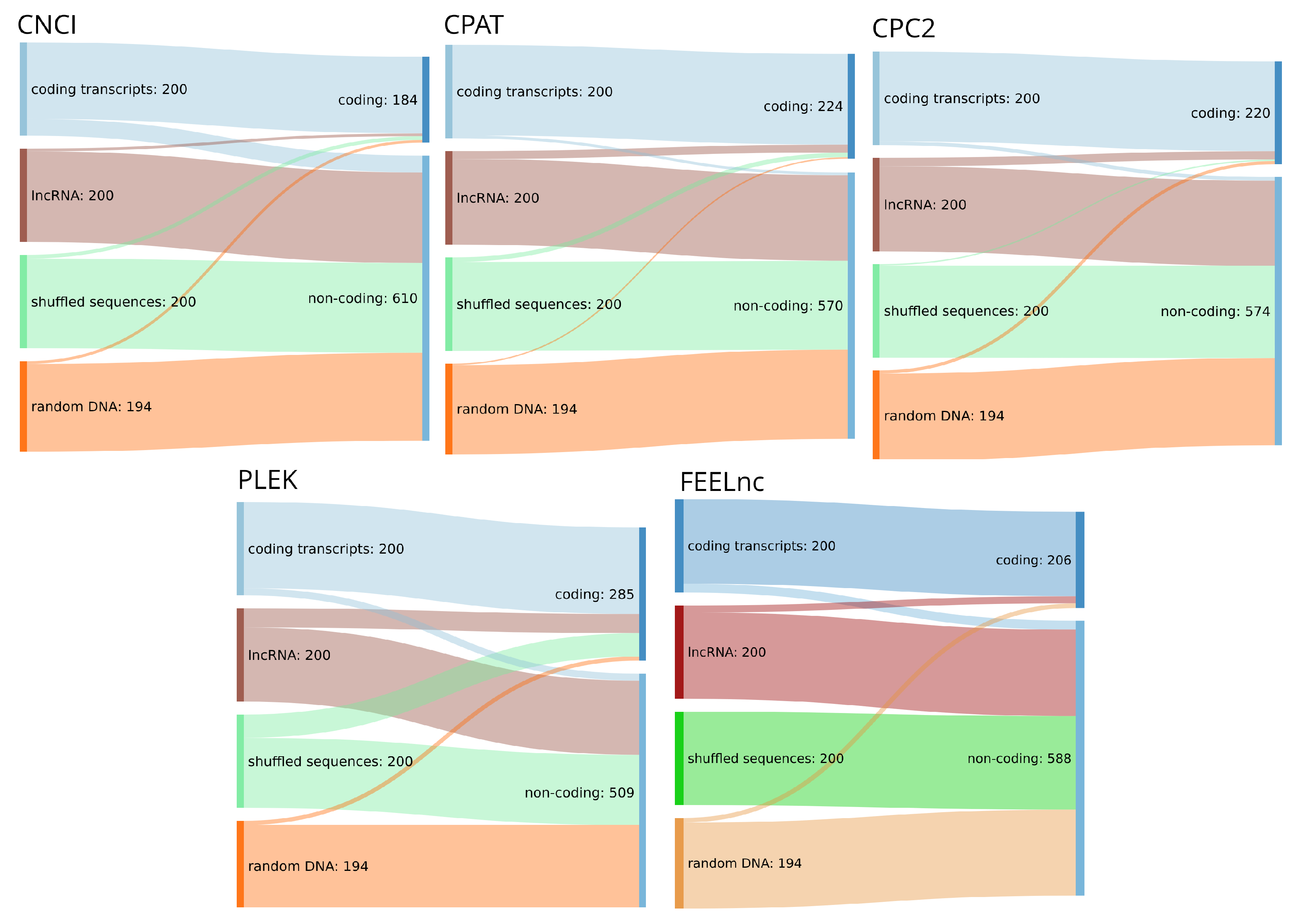
4. Mini Benchmark on Human Transcripts and Randomly Chosen Genomic Sequences
5. Discussion
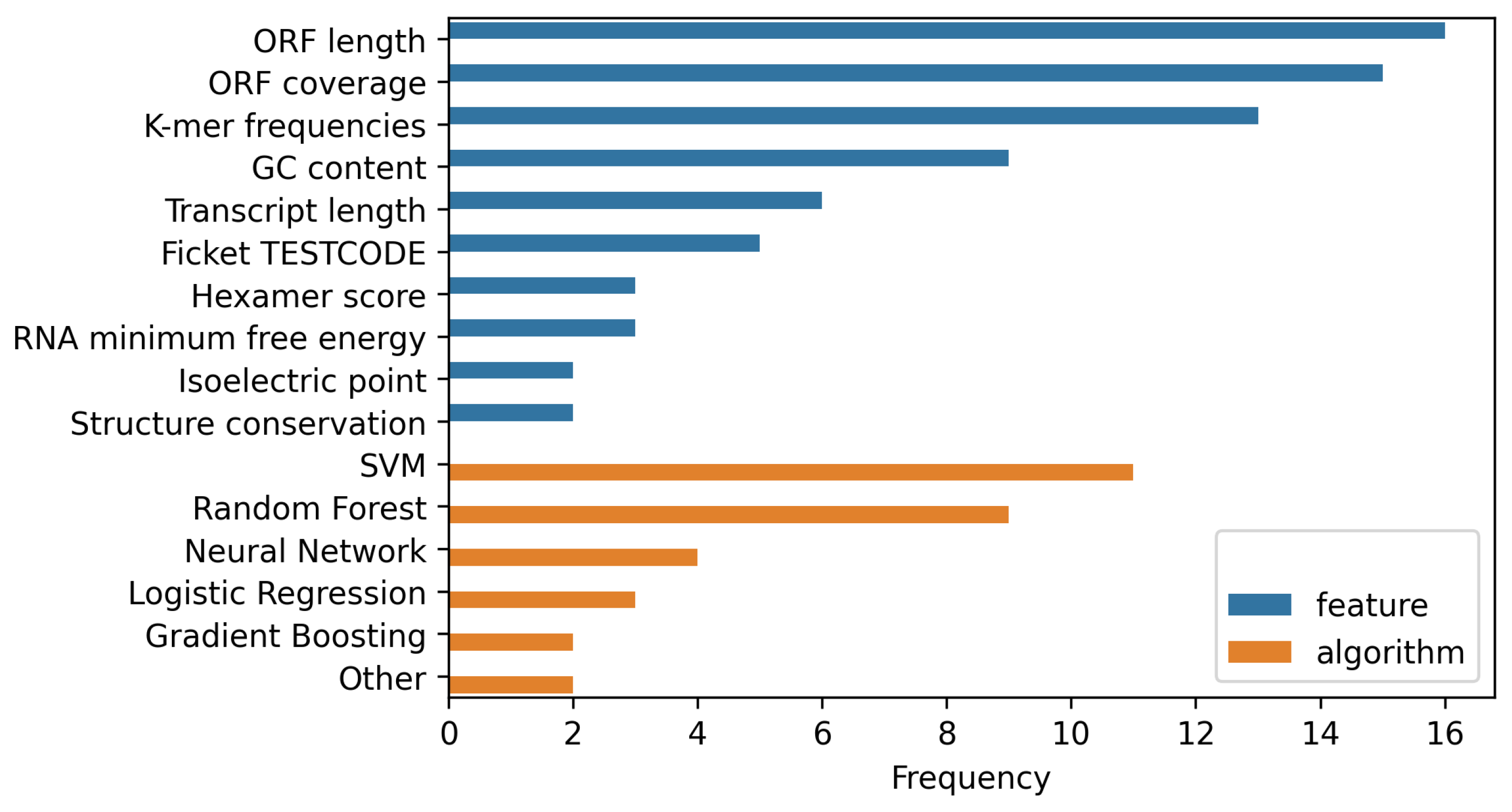
5.1. Classification Accuracy in the Context of Used Features
5.2. Comparing Performance of lncRNA and Coding Potential Classifiers
5.3. Functional Classification of lncRNA
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.C.; Ponting, C.P. Intergenic lncRNAs and the evolution of gene expression. Curr. Opin. Genet. Dev. 2014, 27, 48–53. [Google Scholar] [CrossRef]
- Yang, G.; Lu, X.; Yuan, L. LncRNA: A link between RNA and cancer. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014, 1839, 1097–1109. [Google Scholar] [CrossRef]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354. [Google Scholar] [PubMed]
- Zhang, R.; Xia, L.Q.; Lu, W.W.; Zhang, J.; Zhu, J.S. LncRNAs and cancer. Oncol. Lett. 2016, 12, 1233–1239. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Luo, P.; Wang, Q.; Ye, Y.; Wang, B. lncRNA PVT1 in cancer: A review and meta-analysis. Clin. Chim. Acta 2017, 474, 1–7. [Google Scholar] [CrossRef]
- Amin, N.; McGrath, A.; Chen, Y.P.P. Evaluation of deep learning in non-coding RNA classification. Nat. Mach. Intell. 2019, 1, 246–256. [Google Scholar] [CrossRef]
- Cao, Y.; Geddes, T.A.; Yang, J.Y.H.; Yang, P. Ensemble deep learning in bioinformatics. Nat. Mach. Intell. 2020, 2, 500–508. [Google Scholar] [CrossRef]
- McDonel, P.; Guttman, M. Approaches for understanding the mechanisms of long noncoding RNA regulation of gene expression. Cold Spring Harb. Perspect. Biol. 2019, 11, a032151. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Liang, Y.; Li, Y.; Du, W. Long noncoding RNA identification: Comparing machine learning based tools for long noncoding transcripts discrimination. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- Saghatelian, A.; Couso, J.P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat. Chem. Biol. 2015, 11, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Martinez, T.F.; Chu, Q.; Donaldson, C.; Tan, D.; Shokhirev, M.N.; Saghatelian, A. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 2020, 16, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.A.; Kohajda, T.; Findeiß, S.; Stadler, P.F.; Washietl, S.; Kellis, M.; von Bergen, M.; Kalkhof, S. Optimization of parameters for coverage of low molecular weight proteins. Anal. Bioanal. Chem. 2010, 398, 2867–2881. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, C. Coding or Noncoding, the Converging Concepts of RNAs. Front. Genet. 2019, 2019, 496. [Google Scholar] [CrossRef]
- Findeiß, S.; Engelhardt, J.; Prohaska, S.J.; Stadler, P.F. Protein-coding structured RNAs: A computational survey of conserved RNA secondary structures overlapping coding regions in drosophilids. Biochimie 2011, 93, 2019–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Kirk, J.M.; Kim, S.O.; Inoue, K.; Smola, M.J.; Lee, D.M.; Schertzer, M.D.; Wooten, J.S.; Baker, A.R.; Sprague, D.; Collins, D.W.; et al. Functional classification of long non-coding RNAs by k-mer content. Nat. Genet. 2018, 50, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, Y.; Ma, Q.; Xu, Y.; Zhang, Y.; Du, W.; Wang, C.; Li, Y. LncFinder: An integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief. Bioinform. 2019, 20, 2009–2027. [Google Scholar] [CrossRef]
- Pozzoli, U.; Menozzi, G.; Fumagalli, M.; Cereda, M.; Comi, G.P.; Cagliani, R.; Bresolin, N.; Sironi, M. Both selective and neutral processes drive GC content evolution in the human genome. BMC Evol. Biol. 2008, 8, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fickett, J.W. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 1982, 10, 5303–5318. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Fickett, J.W.; Tung, C.S. Assessment of protein coding measures. Nucleic Acids Res. 1992, 20, 6441–6450. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Simm, S.; Einloft, J.; Mirus, O.; Schleiff, E. 50 years of amino acid hydrophobicity scales: Revisiting the capacity for peptide classification. Biol. Res. 2016, 49, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowski, L.P. IPC—Isoelectric Point Calculator. Biol. Direct 2016, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, M.C.; Bailey, T.L.; Kasukawa, T.; Mignone, F.; Kummerfeld, S.K.; Madera, M.; Sunkara, S.; Furuno, M.; Bult, C.J.; Quackenbush, J.; et al. Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biol. 2006, 3, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Sun, Y.Z.; Guan, N.N.; Qu, J.; Huang, Z.A.; Zhu, Z.X.; Li, J.Q. Computational models for lncRNA function prediction and functional similarity calculation. Brief. Funct. Genom. 2019, 18, 58–82. [Google Scholar] [CrossRef]
- Lin, M.F.; Kheradpour, P.; Washietl, S.; Parker, B.J.; Pedersen, J.S.; Kellis, M. Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res. 2011, 21, 1916–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, A.R.; Findeiß, S.; Washietl, S.; Hofacker, I.L.; Stadler, P.F. RNAz 2.0: Improved noncoding RNA detection. In Biocomputing 2010; World Scientific: Singapore, 2010; pp. 69–79. [Google Scholar] [CrossRef]
- Yao, Z.; Weinberg, Z.; Ruzzo, W.L. CMfinder—A covariance model based RNA motif finding algorithm. Bioinformatics 2006, 22, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.F.; Jungreis, I.; Kellis, M. PhyloCSF: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef] [PubMed]
- Pockrandt, C.; Steinegger, M.; Salzberg, S. PhyloCSF++: A fast and user-friendly implementation of PhyloCSF with annotation tools. Bioinformatics 2021, btab756. [Google Scholar] [CrossRef]
- Washietl, S. Prediction of structural noncoding RNAs with RNAz. In Comparative Genomics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 503–525. [Google Scholar]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Bioinformatics 1993, 9, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washietl, S.; Findeiß, S.; Müller, S.; Kalkhof, S.; von Bergen, M.; Hofacker, I.L.; Stadler, P.F.; Goldman, N. RNAcode: Robust prediction of protein coding regions in comparative genomics data. RNA 2011, 17, 578–594. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Tripathi, R.; Patel, S.; Kumari, V.; Chakraborty, P.; Varadwaj, P.K. DeepLNC, a long non-coding RNA prediction tool using deep neural network. Netw. Model. Anal. Health Inform. Bioinform. 2016, 5, 21. [Google Scholar] [CrossRef]
- Volders, P.J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Ito, E.A.; Katahira, I.; Vicente, F.F.d.R.; Pereira, L.F.P.; Lopes, F.M. BASiNET—BiologicAl Sequences NETwork: A case study on coding and non-coding RNAs identification. Nucleic Acids Res. 2018, 46, e96. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Contreras-Moreira, B.; De Silva, N.; Maslen, G.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Barba, M.; Bolser, D.M.; Cambell, L.; et al. Ensembl Genomes 2020—Enabling non-vertebrate genomic research. Nucleic Acids Res. 2020, 48, D689–D695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Chen, X.; Jiang, P.; Song, X.; Wang, H.; Sun, H. iSeeRNA: Identification of long intergenic non-coding RNA transcripts from transcriptome sequencing data. BMC Genom. 2013, 14, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Haussler, D. Phylogenetic hidden Markov models. In Statistical Methods in Molecular Evolution; Springer: Berlin/Heidelberg, Germany, 2005; pp. 325–351. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Guo, J.C.; Fang, S.S.; Wu, Y.; Zhang, J.H.; Chen, Y.; Liu, J.; Wu, B.; Wu, J.R.; Li, E.M.; Xu, L.Y.; et al. CNIT: A fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res. 2019, 47, W516–W522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Pian, C.; Zhang, G.; Chen, Z.; Chen, Y.; Zhang, J.; Yang, T.; Zhang, L. LncRNApred: Classification of long non-coding RNAs and protein-coding transcripts by the ensemble algorithm with a new hybrid feature. PLoS ONE 2016, 11, e0154567. [Google Scholar] [CrossRef] [PubMed]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49, D165–D171. [Google Scholar] [CrossRef]
- Baek, J.; Lee, B.; Kwon, S.; Yoon, S. LncRNAnet: Long non-coding RNA identification using deep learning. Bioinformatics 2018, 34, 3889–3897. [Google Scholar] [CrossRef]
- LeCun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Yang, L.; Zhou, M.; Xie, H.; Zhang, C.; Wang, M.D.; Zhu, H. LncADeep: An ab initio lncRNA identification and functional annotation tool based on deep learning. Bioinformatics 2018, 34, 3825–3834. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.; Hu, G.; Zhu, H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinform. 2013, 14, S12. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Yin, H.; Li, B.; Yu, C.; Wang, F.; Xu, X.; Cao, J.; Bao, Y.; Wang, L.; Abbasi, A.A.; et al. Characterization and identification of long non-coding RNAs based on feature relationship. Bioinformatics 2019, 35, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.; Shuttleworth, J.; Yang, J.; Taramonli, S.; England, M. PLIT: An alignment-free computational tool for identification of long non-coding RNAs in plant transcriptomic datasets. Comput. Biol. Med. 2019, 105, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Liu, H.; Zhang, L.; Meng, J. lncRScan-SVM: A tool for predicting long non-coding RNAs using support vector machine. PLoS ONE 2015, 10, e0139654. [Google Scholar] [CrossRef]
- Liu, J.; Gough, J.; Rost, B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet. 2006, 2, e29. [Google Scholar] [CrossRef] [Green Version]
- Slater, G. Algorithms for the Analysis of ESTs. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 1998. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, T.U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Arrial, R.T.; Togawa, R.C.; de M Brigido, M. Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: Case study of the pathogenic fungus Paracoccidioides brasiliensis. BMC Bioinform. 2009, 10, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achawanantakun, R.; Chen, J.; Sun, Y.; Zhang, Y. LncRNA-ID: Long non-coding RNA IDentification using balanced random forests. Bioinformatics 2015, 31, 3897–3905. [Google Scholar] [CrossRef] [Green Version]
- Kozak, M. Initiation of translation in prokaryotes and eukaryotes. Gene 1999, 234, 187–208. [Google Scholar] [CrossRef]
- Xu, H.; Wang, P.; Fu, Y.; Zheng, Y.; Tang, Q.; Si, L.; You, J.; Zhang, Z.; Zhu, Y.; Zhou, L.; et al. Length of the ORF, position of the first AUG and the Kozak motif are important factors in potential dual-coding transcripts. Cell Res. 2010, 20, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.; Khemka, N.; Rajkumar, M.S.; Garg, R.; Jain, M. PLncPRO for prediction of long non-coding RNAs (lncRNAs) in plants and its application for discovery of abiotic stress-responsive lncRNAs in rice and chickpea. Nucleic Acids Res. 2017, 45, e183. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, C.M.; Weretilnyk, E.A.; Golding, G.B. Prediction of plant lncRNA by ensemble machine learning classifiers. BMC Genom. 2018, 19, 316. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xu, Z.; Hu, B.; Lu, Z.J. COME: A robust coding potential calculation tool for lncRNA identification and characterization based on multiple features. Nucleic Acids Res. 2017, 45, e2. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Hofacker, I.L.; Stadler, P.F. Local RNA base pairing probabilities in large sequences. Bioinformatics 2006, 22, 614–615. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. AMB 2011, 6, 26. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, X.; Zhang, G.; Li, W.; Liu, F.; Liu, S.; Zhang, W. PredLnc-GFStack: A global sequence feature based on a stacked ensemble learning method for predicting lncRNAs from transcripts. Genes 2019, 10, 672. [Google Scholar] [CrossRef] [Green Version]
- Pyfrom, S.C.; Luo, H.; Payton, J.E. PLAIDOH: A novel method for functional prediction of long non-coding RNAs identifies cancer-specific LncRNA activities. BMC Genom. 2019, 20, 137. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Liu, S.; Yang, Z.; Zhao, X.; Deng, Y.; Zhang, G.; Pang, J.; Zhao, C.; Zhang, W. A Systematic Review of Computational Methods for Predicting Long Noncoding RNAs. Brief. Funct. Genom. 2021, 20, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Gatter, T.; Stadler, P.F. Ryūtō: Improved multi-sample transcript assembly for differential transcript expression analysis. Bioinformatics 2021. [Google Scholar] [CrossRef]
- Long, H.; Sung, W.; Kucukyildirim, S.; Williams, E.; Miller, S.F.; Guo, W.; Patterson, C.; Gregory, C.; Strauss, C.; Stone, C.; et al. Evolutionary determinants of genome-wide nucleotide composition. Nat. Ecol. Evol. 2018, 2, 237–240. [Google Scholar] [CrossRef]
- Reichenberger, E.R.; Rosen, G.; Hershberg, U.; Hershberg, R. Prokaryotic nucleotide composition is shaped by both phylogeny and the environment. Genome Biol. Evol. 2015, 7, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Katti, M.V.; Ranjekar, P.K.; Gupta, V.S. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 2001, 18, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhang, L.; Jia, L.; Duan, Y.; Li, Y.; Bao, L.; Sha, N. Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation. PLoS ONE 2014, 9, e100893. [Google Scholar]
- Ramos, A.D.; Andersen, R.E.; Liu, S.J.; Nowakowski, T.J.; Hong, S.J.; Gertz, C.C.; Salinas, R.D.; Zarabi, H.; Kriegstein, A.R.; Lim, D.A. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 2015, 16, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [Green Version]
- Volders, P.J.; Helsens, K.; Wang, X.; Menten, B.; Martens, L.; Gevaert, K.; Vandesompele, J.; Mestdagh, P. LNCipedia: A database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013, 41, D246–D251. [Google Scholar] [CrossRef] [Green Version]
- Liakos, K.G.; Busato, P.; Moshou, D.; Pearson, S.; Bochtis, D. Machine learning in agriculture: A review. Sensors 2018, 18, 2674. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Pan, Y.; Pan, Y.; Wang, O. MNX1-AS1 is a functional oncogene that induces EMT and activates the AKT/mTOR pathway and MNX1 in breast cancer. Cancer Manag. Res. 2019, 11, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhai, D.; Huang, Q.; Chen, H.; Zhang, Z.; Tan, Q. LncRNA DCST1-AS1 accelerates the proliferation, metastasis and autophagy of hepatocellular carcinoma cell by AKT/mTOR signaling pathways. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6091–6104. [Google Scholar] [PubMed]
- Bonidia, R.P.; de Leon Ferreira de Carvalho, A.C.P.; Paschoal, A.R.; Sanches, D.S. Selecting the most relevant features for the identification of long non-coding RNAs in plants. In Proceedings of the 2019 8th Brazilian Conference on Intelligent Systems (BRACIS), Salvador, Brazil, 15–18 October 2019; pp. 539–544. [Google Scholar]
- Liu, C.; Lin, J. Long noncoding RNA ZEB1-AS1 acts as an oncogene in osteosarcoma by epigenetically activating ZEB1. Am. J. Transl. Res. 2016, 8, 4095. [Google Scholar]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafà, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Xu, M.; Shi, H.; Gao, X.; Liang, P. Genome-wide identification of lncRNAs associated with chlorantraniliprole resistance in diamondback moth Plutella xylostella (L.). BMC Genom. 2017, 18, 380. [Google Scholar] [CrossRef]
- Volders, P.J.; Verheggen, K.; Menschaert, G.; Vandepoele, K.; Martens, L.; Vandesompele, J.; Mestdagh, P. An update on LNCipedia: A database for annotated human lncRNA sequences. Nucleic Acids Res. 2015, 43, D174–D180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, L. Screening for lncRNA function. Nat. Rev. Genet. 2017, 18, 70. [Google Scholar] [CrossRef]
- Wang, C.; Wang, L.; Ding, Y.; Lu, X.; Zhang, G.; Yang, J.; Zheng, H.; Wang, H.; Jiang, Y.; Xu, L. LncRNA structural characteristics in epigenetic regulation. Int. J. Mol. Sci. 2017, 18, 2659. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.T.; He, J.; Liang, Q.; Ren, L.L.; Yan, T.T.; Yu, T.C.; Tang, J.Y.; Bao, Y.J.; Hu, Y.; Lin, Y.; et al. LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov. 2016, 6, 784–801. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef]
- Aznaourova, M.; Schmerer, N.; Schmeck, B.; Schulte, L.N. Disease-causing mutations and rearrangements in long non-coding RNA gene loci. Front. Genet. 2020, 11, 1485. [Google Scholar] [CrossRef]
- Henshall, D.C. Epigenetics and noncoding RNA: Recent developments and future therapeutic opportunities. Eur. J. Paediatr. Neurol. 2020, 24, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Fallmann, J.; Walter, M.E.M.T.; Stadler, P.F. Are spliced ncRNA Host Genes distinct classes of lncRNAs? Theory Biosci. 2020, 139, 349–359. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tool | Year | Algorithm | Species | Features | Performance | Mean Citations per Year |
|---|---|---|---|---|---|---|
| CONC | 2006 | SVM | Eukaryotes (both protein-coding and non-coding genes) | peptide length, amino acid composition, predicted secondary structure content, mean hydrophobicity, percentage of residues exposed to solvent, sequence compositional entropy, number of homologues, alignment entropy | 10-fold CV on protein-coding: F1-score: 97.4% ☼ Precision: 97.1% ☼ Recall: 97.8% ◙ On non-coding: F1-score: 94.5% ☼ Precision: 95.2% ☼ Recall: 93.8% | 12.4 |
| CPC | 2007 | SVM | Eukaryotes (both protein-coding and non-coding genes) | ORF features (quality, coverage, integrity), number of BLASTX hits, hit score, frame score | 10-fold CV: 95.77% ☼ Accuracy on Rfam database (non-coding): 98.62% ☼ RNADB (non-coding): 91.5% ☼ EMBL cds (protein-coding): 99.08% ◙ Accuracy in lncRNA detection: 76.2% | 131.8 |
| PORTRAIT | 2009 | SVM | Species neutral, case study on Paracoccidioides brasiliensis and 5 other fungi | ORF length, isoelectric point, hydropathy, compositional entropy | Accuracy: 91.9% ☼ Specificity: 95% ☼ Sensitivity: 86.4% ☼ | 10.2 |
| CNCI | 2013 | SVM | Vertebrates, plants, orangutan | adjacent nucleotide triplets, sequence score, codon-bias, most-like CDS (MLCDS), length-percentage, score-distance | 10-fold CV accuracy on human: 97.3% ◙ Minimum average error for vertebrates < 0.1 ☼ Plants: 0.24 | 111.3 |
| CPAT | 2013 | Logistic regression | human | ORF length, ORF to transcript length ratio, Fickett score, hexamer usage bias | 10-fold accuracy: 99% ☼ Precision: 96% | 135.6 |
| iSeeRNA | 2013 | SVM | human, mouse | frequency of six k-mers (GC, CT, TAG, TGT, ACG, TCG), conservation score, ORF length and proportion | Accuracy in human lncRNA detection: 96.1% ☼ Mouse: 94.2% ◙ Accuracy in human protein-coding gene detection: 94.7% ☼ Mouse: 92.7% | 19.5 |
| PLEK | 2014 | SVM | 11 vertebrates | k-mer frequency (for k = [1,5]) | 10-fold CV accuracy: 95.6% | 50.5 |
| lncRScan-SVM | 2015 | SVM | human, mouse | sum of lengths of exons, frequency of exons, mean exon length, standard deviation of stop codon frequency, txCdsPredict | Two test sets created based on (i) random protein-coding and lncRNA sequences and (ii) only dissimilar sequences. Accuracy on set A for human: 91.54% ☼ Mouse: 92.21% ◙ On set B for human: 91.45% ☼ Mouse: 92.2% ◙ MCC on set A for human: 83.17% ☼ Mouse: 84.59% ◙ On set B for human: 82.99% ☼ Mouse: 84.69% ◙ AUC on set A for human: 96.39% ☼ Mouse: 96.62% ◙ On set B for human: 96.39% ☼ Mouse: 96.64% ◙ | 13.2 |
| LncRNA-ID | 2015 | Random forests | human, mouse | ORF related features, ribosomal interaction related features, protein conservation scores | Specificity on human: 95.28% ☼ Mouse: 92.1% ◙ Recall on human: 96.28% ☼ Mouse: 94.45% ◙ Accuracy on human: 95.78% ☼ Mouse: 93.28% | 12.7 |
| COME | 2016 | Random forest | human, mouse, nematode, fruit fly, arabidopsis | GC content, DNA sequence conservation, protein conservation, polyA abundance, RNA secondary structure conservation, ORF score, expression specificity score | Accuracy: human (93.7%), arabidopsis (98.3%), mouse (89.8%), nematode (98.9%), fruit fly (98.4%) | 16.2 |
| DeepLNC | 2016 | Deep neural network | human | k-mer combinations (for k = [2,5]) | 10-fold CV accuracy: 98.07% ☼ MCC: 96% ☼ Recall: 98.98% ☼ Precision: 97.14% ☼ AUC: 99.3% | 12.4 |
| FEELnc | 2017 | Random forests | human, mouse | ORF features (coverage, length), sequence length, coding potential score, k-mer score based on frequency | Accuracy for human: 91.9% ☼ Mouse: 93.9% ◙ Sensitivity for human: 92.3% ☼ Mouse: 93.8% ◙ Specificity for human: 91.5% ☼ Mouse: 94.1% ◙ F score for human: 91.9% ☼ Mouse: 95.6% ◙ MCC for human: 83.8% ☼ Mouse: 85.6% | 49.5 |
| CPC2 | 2017 | Random forest | Species neutral, trained and tested on animals and plants (both protein-coding and non-coding genes) | ORF features (quality, coverage, integrity), Fickett score, isoelectric point | Accuracy: 96.1% ☼ Specificity: 97% ☼ Recall: 95.2% ◙ Accuracy in lncRNA detection: 94.2% | 97.3 |
| PlncPRO | 2017 | Random forest | plants | 64 k-mer frequencies, ORF coverage, ORF score, BLASTX: hits, significance, total bit score, frame entropy | 13.8 | |
| lncRNAnet | 2018 | Convolutional neural network, recurrent neural network | human, mouse | sequence, ORF features (length, coverage, indicator) | 5-fold accuracy: 99% ◙ Accuracy on human: 91.79% ☼ Mouse: 91.83% ◙ Specificity on human: 87.66% ☼ Mouse: 89.03% ◙ Sensitivity on human: 95.91% ☼ Mouse: 94.63% ◙ AUC on human: 96.72% ☼ Mouse: 96.67% ↯Also available are test results on 11 different species and on experimental NGS data. | 22 |
| lncADeep | 2018 | Deep belief network | human, mouse | ORF features (length, coverage, hexamer score of longest ORF, entropy density profile), UTR coverage, GC content of UTRs, Fickett score, HMMER index | Precision for lncRNA detection from full-length mRNA transcripts: 97.2% ☼ Recall: 98.1% ☼ Average harmonic mean: 97.7% ◙ Precision for lncRNA detection from both full and partial-length mRNA transcripts: 94.5% ☼ Recall: 93.8% ☼ Average harmonic mean: 94.2% ◙ Precision for lncRNA detection from partial-length mRNA transcripts: 90.3% ☼ Recall: 93.8% ☼ Average harmonic mean: 92% | 22.6 |
| LncFinder | 2018 | SVM | Trained on human, tested on human, mouse, wheat, zebrafish, chicken | genomic distance to lncRNA, genomic distance to protein-coding transcript, distance ratio, EIIP value | 10-fold CV accuracy: 96.87% | 17.6 |
| BASiNET | 2018 | Decision tree on complex networks | datasets from PLEK and CPC2 | average shortest path, average betweenness centrality, average degree, assortativity, maximum degree, minimum degree, clustering coefficient, motif frequency | 8.6 | |
| CREMA | 2018 | Random forest | human, mouse, rice, arabidopsis | length, GC content, hexamer score, alignment identity, ratio of alignment length and mRNA length, ratio of alignment length and ORF length, transposable elements, sequence divergence from transposable element, ORF length, Ficket score | 11 | |
| CNIT | 2019 | SVM | 11 animal species, 26 plant species | max_score of MLCDS, standard deviation of MLCDS scores and MLCDS lengths, frequency of 64 codons | Accuracy on human: 98% ☼ Mouse: 95% ☼ Zebrafish: 93% ☼ Fruit fly: 93% ☼ arabidopsis: 98% | 20.5 |
| PLIT | 2019 | Random forest | plants: arabidopsis, soy bean, rice, tomato, sorghum, vine grape, maize | transcript length, GC content, Ficket-score, hexamer score, maximum ORF length, ORF coverage, mean ORF coverage, codon bias | AUC: 93.3 % for everything except S. bicolor (75%) and arabidopsis (85%) | 8.5 |
| LGC | 2019 | Feature relationship | human, mouse, zebrafish, nematode, rice, tomato | GC content, ORF length, coding potential score | Accuracy, 10 fold cross-validation: human (94.5%), mouse (93.6%), zebrafish (88.4%), nematode (93.3%), tomato (93.3%), rice (96.3%) | 12.5 |
| Tool | Year | Algorithm | Citations | Species |
|---|---|---|---|---|
| CPC | 2007 | SVM | 1857 | Eukaryotes (protein-coding and non-coding transcripts) |
| CNCI | 2013 | SVM | 898 | Vertebrates, plants, orangutan |
| CPAT | 2013 | logistic regression | 1105 | human |
| PLEK | 2014 | SVM | 359 | 11 vertebrate species |
| CPC2 | 2017 | Random forest | 406 | Eukaryotes (protein-coding and non-coding transcripts) |
| FEELnc | 2017 | Random Forest | 198 | human, mouse |
| Tool | Sensitivity | Specificity | Accuracy | Precision |
|---|---|---|---|---|
| CNCI | 0.82 | 0.97 | 0.895 | 0.96 |
| CPAT | 0.97 | 0.92 | 0.94 | 0.92 |
| PLEK | 0.93 | 0.79 | 0.86 | 0.82 |
| CPC2 | 0.96 | 0.91 | 0.94 | 0.91 |
| FEELnc | 0.91 | 0.93 | 0.92 | 0.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klapproth, C.; Sen, R.; Stadler, P.F.; Findeiß, S.; Fallmann, J. Common Features in lncRNA Annotation and Classification: A Survey. Non-Coding RNA 2021, 7, 77. https://doi.org/10.3390/ncrna7040077
Klapproth C, Sen R, Stadler PF, Findeiß S, Fallmann J. Common Features in lncRNA Annotation and Classification: A Survey. Non-Coding RNA. 2021; 7(4):77. https://doi.org/10.3390/ncrna7040077
Chicago/Turabian StyleKlapproth, Christopher, Rituparno Sen, Peter F. Stadler, Sven Findeiß, and Jörg Fallmann. 2021. "Common Features in lncRNA Annotation and Classification: A Survey" Non-Coding RNA 7, no. 4: 77. https://doi.org/10.3390/ncrna7040077
APA StyleKlapproth, C., Sen, R., Stadler, P. F., Findeiß, S., & Fallmann, J. (2021). Common Features in lncRNA Annotation and Classification: A Survey. Non-Coding RNA, 7(4), 77. https://doi.org/10.3390/ncrna7040077